Medical device manufacturers require systematic procedures to report risks or problems associated with the use of their devices at an early stage to the competent authority so that appropriate action is taken. Reporting of such incidents improve safety of patients and users and also reduce reoccurrence of such incidents.
On 30 January 2024, Medical Device Coordination Group publishes “Guidance on the vigilance system for CE-marked devices DSVG 00 – Device Specific Vigilance Guidance (DSVG) Template” and specific device vigilance guidance document on:
- Device for Cardiac Ablation, MDCG -1-1 DSVG 01
- Coronary Stents and associated delivery system, MDCG 2024-1-2 DSVG 02
- Cardiac Implantable Electronic Devices (CIEDS) MDCG 2024-1-3, DSVG 03
- Breast Implants MDCG-1-4, DSVG 04
Scope
The Device Specific Vigilance Guidance (DSVG) aims to harmonise vigilance reporting and provide manufacturers with guidance on effectively reporting incidents to the relevant competent authority. These guidelines are to be utilized in conjunction with Regulation (EU) 2017/745 on medical devices (MDR), specifically focusing on Article 87 and 88 for a comprehensive understanding of vigilance reporting obligations.
This guidance document provides following types of vigilance reporting for specific types of devices:
- Individual serious incident
- Periodic Summary Reporting
- Trend Reporting
Individual serious incident
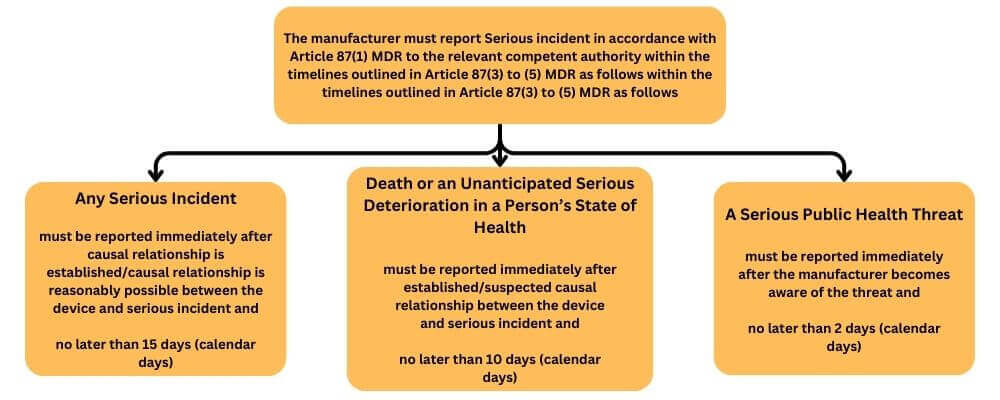
In accordance with Article 87 MDR all individual serious incidents should be reported to the competent authority. This also applies in cases where the manufacturer is unclear whether the incident is reportable or needs clarification for the root cause of the device. The manufacturer must report a serious incident to the competent authority in the given timeframe.
serious incident’ means any incident that directly or indirectly led, might have led or might lead to any of the following:
- the death of a patient, user or other person,
- the temporary or permanent serious deterioration of a patient’s, user’s or other person’s state of health,
- a serious public health threat;
Reporting period begins on the day after the awareness date of potentially serious incident (manufacturer awareness date (day = 0)
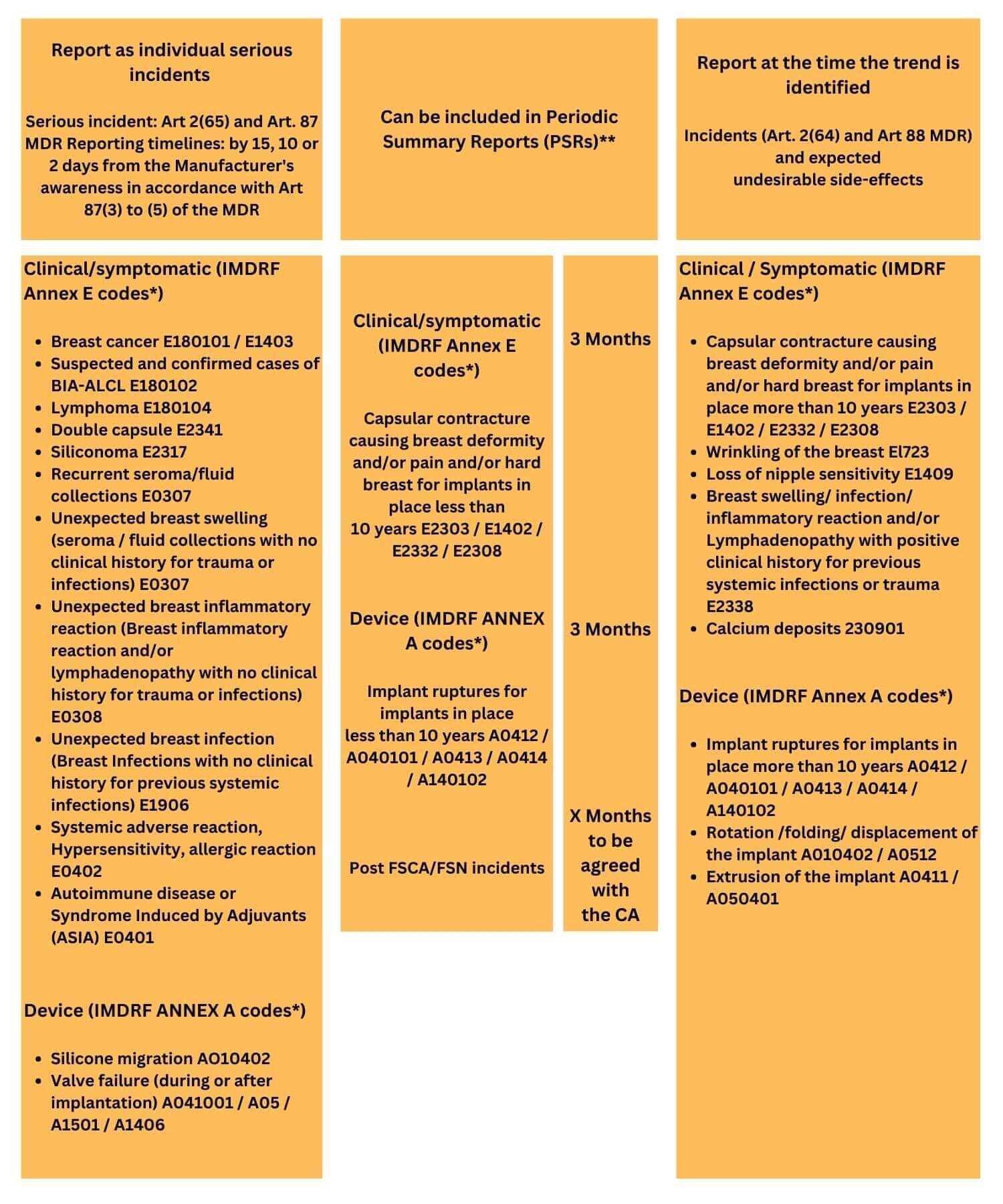
Periodic Summary Reporting
In accordance with the Article 87(9) MDR manufacturer can report similar serious incidents involving the same device or device type to the competent authority in a consolidated way using periodic summary report. The format, content and frequency of reporting periodic summary reports should be coordinated with the Competent Authority.
Trend reporting
In accordance with Article 88 MDR manufacture requires to report to a competent authority when there is a substantial rise in the frequency or severity of incidents (not serious incidents) or anticipated undesirable side effects linked to their device. These incidents may significantly affect the benefit-risk analysis, making the observation of trends an indicator of a potential shift in the risk-benefit ratio.
IMDRF Terminologies for Categorised Adverse Event Reporting
● For reporting device specific serious incidents, IMDRF Annex A and IMDRF Annex E codes were used.
● IMDRF Annex A code describes medical device problem without going into the details of cause of the problem or failure.
● IMDRF Annex E codes describe patients conditions (Clinical signs and symptoms) related to the medical device adverse events without using diagnostic specifies.
Example – Breast Implants
MDCG 2024-1-4 provides guidance specific to vigilance reporting for breast implants. This guideline encompasses the specific incidents that necessitate reporting, including individual serious incidents, Periodic Summary Reporting, and Trend Reporting, with illustrative examples (does not constitute an exhaustive list).
In conclusion, this guidance document facilitates the smooth transition of regulatory requirements for vigilance reporting by providing valuable assistance in navigating the associated regulatory landscape.
References
1. Regulation (EU) 2017/745 of the European Parliament and of the Council of 5 April 2017 on medical devices, amending Directive 2001/83/EC, Regulation (EC) No 178/2002 and Regulation (EC) No 1223/2009 and repealing Council Directives 90/385/EEC and 93/42/EEC
2. IMDRF terminologies for categorized Adverse Event Reporting (AER): terms, terminology structure and codes
3. MDCG 2024-1(1-4): Guidance on the vigilance system for CE-marked devices