Post-market surveillance (PMS) is a system that gives continuous feedback about the performance of devices on the market in order to maintain a high standard of product quality. It is an essential part of regulatory requirements and the Medical Device Regulation (EU MDR) and In-Vitro Diagnostic Regulation (IVDR) give special importance to gathering clinical and safety-related data after the approval/CE certification process and market access. The data gathered from effective Post-market surveillance acts as critical input into the update of many technical documents like Clinical evaluation, Risk analysis, usability, etc.
Manufacturers of MD and IVD devices must have an appropriate system for gaining and reviewing experience in the post-production phase from the range of devices he manufactures. NB has to audit to verify the effectiveness of the Post-market surveillance (PMS) system. Such Post-market surveillance systems are an essential part of a manufacturer’s quality assurance system. In most cases, Post-market surveillance (PMS) systems already exist to meet internal company needs, as an integrated part of a manufacturer’s quality system, and/or to meet the requirements of third parties. In the absence of an approved quality system, the manufacturer is still required to have effective Post-market surveillance (PMS) system in place



Post-market surveillance is an integral part of the requirements laid out by ISO 13485. Post-market surveillance can also recognize the new opportunities for improvement associated with the medical device in accordance with ISO 13485. Furthermore, it provides input into the design and development change processes, in accordance with ISO 13485. The Post Market Surveillance (PMS) should be an integral part of the company’s Quality Management System (ISO 13485) to analyze the data on quality, performance, and safety of the medical device throughout its lifecycle.
| ISO 13485-QMS | ISO TR 20416-PMS |
|---|---|
|
|
Relationship between ISO 13485 and Post-market surveillance (PMS)
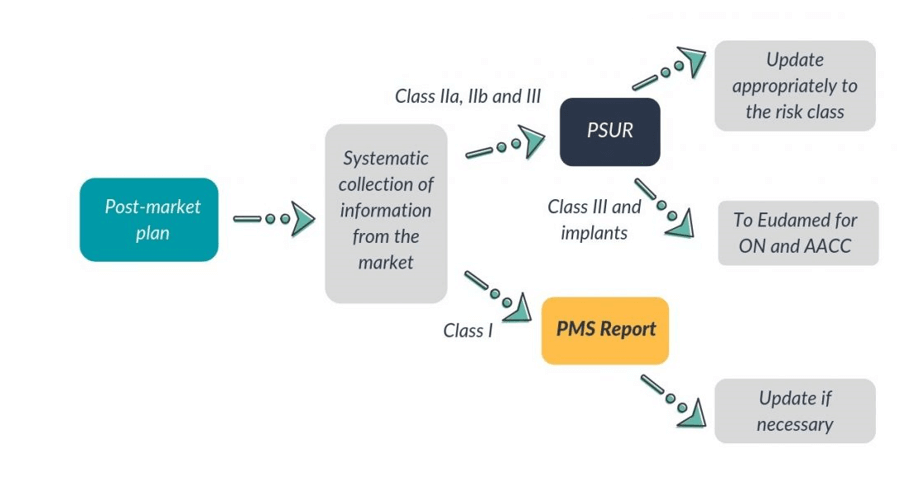
A Post Market Surveillance Plan is a documented strategy of the proactive process for the collection of any required information to evaluate the safety and performance of the medical device released in the market.
The PMS plan contains the requirement of Article 84 & Annex III of EU-MDR to be followed for the collection of data which is directly proportional to the risk associated with the intended use of the medical device.
The post-market surveillance (PMS) plan shall address the collection and utilization of available information, in particular: