The MDR certification landscape continues to evolve as Notified Bodies seek greater consistency in how they interpret and apply Regulation (EU) 2017/745 (MDR). The newly released Team-NB Position Paper (Version 2, June 2026) supports a more harmonized certification approach.
Although the document does not introduce new legal requirements, it provides practical guidance on how certification activities are expected to be conducted throughout the certification lifecycle; from the first interaction with a Notified Body to post-certification surveillance activities.
For manufacturers, this reinforces that MDR certification is not a one-time approval but an ongoing regulatory process that extends beyond certification into post-market activities.
Why This Guidance Matters
What if a well-prepared MDR submission still faces delays due to differing Notified Body expectations?
Variations in application requirements, documentation expectations and assessment approaches can create uncertainty for manufacturers. The Team-NB consensus document seeks to address these challenges by aligning expectations across key stages of the conformity assessment process, supporting more consistent, efficient, and predictable MDR certification.
MDR Certification Is a Multi-Stage Lifecycle Process
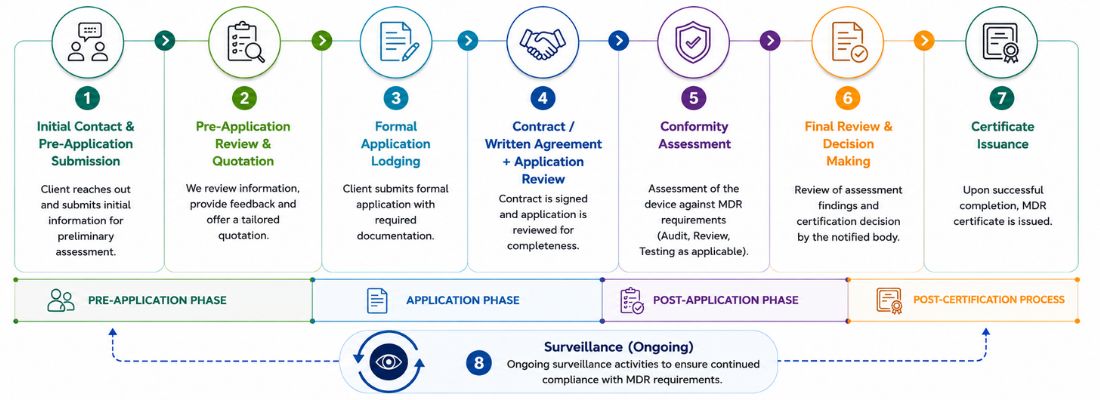
The MDR certification process follows a structured pathway that enables manufacturers to demonstrate compliance with Regulation (EU) 2017/745 before placing devices on the European market.
1. Initial Contact & Pre-Application Submission
Purpose: Establish communication with the Notified Body (NB) and define the certification scope.
- Submission of preliminary device and company information.
- Preliminary review of MDR applicability and device classification.
- Manufacturers or EU Representatives must submit the required Annex A information to enable the Notified Body to prepare a conformity assessment quotation.
2. Pre-Application Review & Quotation Process
Purpose: Enable the NB to evaluate the project scope, complexity, and assessment requirements.
- Verification of MDR scope and device classification.
- Review of manufacturer, site, supplier, and product information.
- Clarification of missing or incomplete information.
- Issuance of a quotation for conformity assessment services.
3. Formal Application Submission
Purpose: Officially initiate the MDR certification process.
- Submission of Quality Management System (QMS) documentation.
- Submission of technical and regulatory documentation.
- Justification of Medical Device intended purpose, qualification and classification.
- Formal lodging of the certification application.
Note: Legacy devices transitioning under MDR may submit a technical documentation plan rather than the complete technical file at this stage.
4. Contract/Written Agreement & Application Review
Purpose: Confirm the application’s completeness and suitability for assessment.
- Establishment of a contract or written agreement.
- Review of application completeness.
- Verification of the selected conformity assessment route.
- Application acceptance or rejection.
5. Conformity Assessment
Purpose: Demonstrate compliance with MDR safety, performance, and quality requirements.
- QMS audits.
- Technical Documentation Assessment (TDAR).
- Clinical Evaluation Assessment (CEAR).
- Product testing and verification, where applicable.
- Additional consultations for specific device categories.
6. Final Review & Certification Decision
Purpose: Confirm that all MDR requirements have been successfully met.
- Independent review of assessment outcomes.
- Verification of corrective actions and non-conformity closure.
- Review of post-market surveillance arrangements.
- Certification approval or refusal.
7. Certificate Issuance
Purpose: Formally certify the device’s compliance with MDR requirements.
- Issuance of the MDR certificate.
- Registration of certification details in EUDAMED.
- Confirmation of compliance with Regulation (EU) 2017/745.
- Authorization to continue placing the device on the EU market.
8. Surveillance
Purpose: Ensure continued compliance throughout the certificate lifecycle.
- Periodic surveillance audits.
- Review of vigilance reports and PSURs.
- Assessment of significant device or QMS changes.
- Technical documentation sampling.
- Unannounced audits, where applicable.
MDR certification is not a one-time event. Manufacturers must continuously demonstrate compliance throughout the validity of the certificate.
Data Readiness: A Critical MDR Requirement
The Team-NB guidance emphasizes that MDR certification starts with strong data readiness. Manufacturers should maintain accurate and up-to-date information on:
- Company and site details
- SRN and PRRC information
- Suppliers and subcontractors
- Device classification, intended purpose, and Basic UDI-DI
- EMDN/MDR codes and sterilization details
- Existing certifications and technical documentation plans
Regulatory Takeaways
The guidance reinforces several important MDR expectations:
- Certification readiness begins before formal application.
- Data quality directly impacts assessment timelines.
- Legacy devices may rely on structured documentation plans during transition.
- Post-market surveillance is a continuous responsibility.
- Effective change management is essential for maintaining compliance.
What Manufacturers Should Do
To improve certification readiness, manufacturers should focus on:
- Strengthening pre-application data quality.
- Confirming device classification and intended purpose.
- Maintaining robust supplier and site documentation.
- Planning MDR technical documentation activities early.
- Strengthening PMS, PMCF, and vigilance processes.
- Implementing formal change control procedures.
Conclusion
While the Team-NB Position Paper does not introduce new MDR requirements, it provides valuable insight into how Notified Bodies are expected to apply them in practice.
The overarching message is clear: successful MDR certification depends on data readiness, proactive planning, and continuous compliance throughout the device lifecycle.
Reference
1. MDR Certification Process (including Pre-application, Application and Post Application phases) – Consensus document