A custom-made device (CMD) is a medical device specifically designed and manufactured for an individual patient or healthcare professional. Unlike mass-produced medical devices, CMDs are created based on a written prescription from a qualified healthcare professional who specifies the device’s design requirements. These devices cater to unique anatomical, physiological, or pathological needs, ensuring a personalized solution that standard devices cannot provide. Examples include prosthetics, orthoses, and dental implants. CMDs are exempt from certain regulatory requirements, such as CE marking, but must still meet quality and safety standards under regulations like EU MDR 2017/745 to ensure patient safety and efficacy.
Medical device regulation for Custom-made devices
In the dynamic world of medical devices, where innovation is constant and patient needs vary widely, custom-made medical devices play a crucial role. These devices are tailored to individual patient’s unique anatomical or physiological characteristics, thereby offering personalized solutions that standard off-the-shelf products often cannot provide. However, the development and use of custom-made medical devices are governed by specific regulations to ensure their safety, efficacy, and quality. In this blog, we’ll explore the regulatory environment pertaining to custom-made medical devices, emphasizing important factors that should be taken into consideration by manufacturers, medical professionals, and patients.
Defining Custom-Made Medical Devices
Before diving into regulations, it’s essential to understand what custom-made medical devices denote.
A ‘custom-made device’ is defined as any device that:
Is intended solely for the use of a particular individual (which could be a patient or healthcare professional);
Is created based on a written request from an authorized healthcare professional, who takes responsibility for providing specific design characteristics even though the design may be developed in consultation with a manufacturer
Is designed to cater to the distinct anatomical, physiological, or pathological attributes of the individual for whom it is intended.
However, mass-produced devices that are adapted to fit the specific needs of any professional user, as well as devices mass-produced through industrial manufacturing methods following written prescriptions from any authorized individual, do not fall under the category of custom-made devices.
Some of the examples includes:
An orthosis made in accordance with a written prescription containing specific design characteristics to aid a person with neuromuscular or musculoskeletal impairment of the lower extremity, such as a Knee Ankle Foot Orthosis (KAFO).
Hand prosthesis intended to replace a lost body part and/or function made in accordance with a written prescription, where the partitioner provides patient specific design characteristics necessary for the manufacturing of the device.
Are the parts, components or materials specifically intended for use in a custom-made device are medical devices in accordance with Regulation 2017/745?
Under Regulation (EU) 2017/745 (MDR), the classification of parts, components, or materials used in custom-made devices depends on their intended purpose. According to the MDR, a medical device is defined as any instrument, apparatus, appliance, software, implant, reagent, material, or similar article intended by the manufacturer to be used for medical purposes, such as diagnosis, prevention, monitoring, treatment, or alleviation of disease.
For custom-made medical devices, manufacturers often use specific parts, components, or materials to ensure that the final product meets the unique needs of a patient. However, not all of these elements are automatically classified as medical devices. The key factor determining their classification is whether they have an independent medical function or are explicitly intended by their manufacturer for medical use.
If a component, part, or material is marketed separately and intended specifically for use in a medical device, it may itself be classified as a medical device under MDR. For example, an implantable joint replacement component intended for integration into a custom prosthetic device could be considered a medical device. Conversely, general-use materials like titanium or medical-grade silicone, which could be used in various applications beyond medical devices, are not classified as medical devices unless the manufacturer specifically designates them for such use.
Manufacturers must carefully evaluate the intended use and regulatory classification of each component to ensure compliance with MDR requirements, particularly regarding safety, quality, and performance.
What are the differences between the requirements of the MDD and the MDR regarding to custom-made devices?
The transition from the Medical Device Directive (MDD) to the Medical Device Regulation (MDR) brought significant changes in regulatory requirements, including stricter oversight of custom-made medical devices. While both frameworks aim to ensure patient safety and product quality, the MDR introduces more comprehensive requirements, enhancing transparency and post-market surveillance.
1. Regulatory Oversight and Definitions:
Under the MDD (93/42/EEC), the definition of custom-made devices was relatively broad, with fewer requirements for manufacturers regarding compliance. However, the MDR (2017/745) refines this definition and establishes stricter documentation and assessment processes, particularly for high-risk Class III custom-made implantable devices.
2. Documentation and Compliance Requirements:
The MDD required manufacturers to prepare a statement confirming that the device was custom-made and compliant with essential requirements. In contrast, the MDR mandates detailed technical documentation (Annex XIII) and conformity assessments (Annex IX) for Class III implantable custom-made devices, ensuring greater regulatory scrutiny.
3. CE Marking and Exemptions:
Both the MDD and MDR exempt custom-made devices from CE marking requirements. However, under the MDR, manufacturers must retain documentation for at least 15 years (Annex XII), ensuring long-term accountability.
4. Unique Device Identification (UDI) and Registration:
MDR introduces the EUDAMED database, requiring registration of custom-made device manufacturers. The UDI system, which applies to standard medical devices, is not mandatory for custom-made devices, maintaining some flexibility for manufacturers.
Regulatory Framework
Custom-made medical devices are subjected to regulatory oversight to ensure patient safety and product quality. The regulations governing these devices typically fall under the broader umbrella of Medical Device Regulations (MDR). However, due to their unique nature, custom-made devices often have distinct regulatory requirements.
To clarify the regulatory requirements for these devices, the Medical Device Coordination Group (MDCG) and the International Medical Device Regulators Forum (IMDRF) prepared the following guidance documents in March 2020 and March 2021, respectively:
The IMDRF wrote the “Personalized Medical Devices – Regulatory Pathways
The MDCG wrote the “MDC 2021-3 Questions and Answers on Custom-Made Devices
In the European Union (EU), custom-made medical devices are regulated under the Medical Devices Regulation (MDR). As per Medical Device Regulation 2017/245, Custom-made devices are tailored to the specific requirements of an individual patient and are exempt from certain regulatory requirements. Also, manufacturers of custom-made devices may demonstrate the requisite expertise by having at least two years of professional experience within a relevant field of manufacturing.
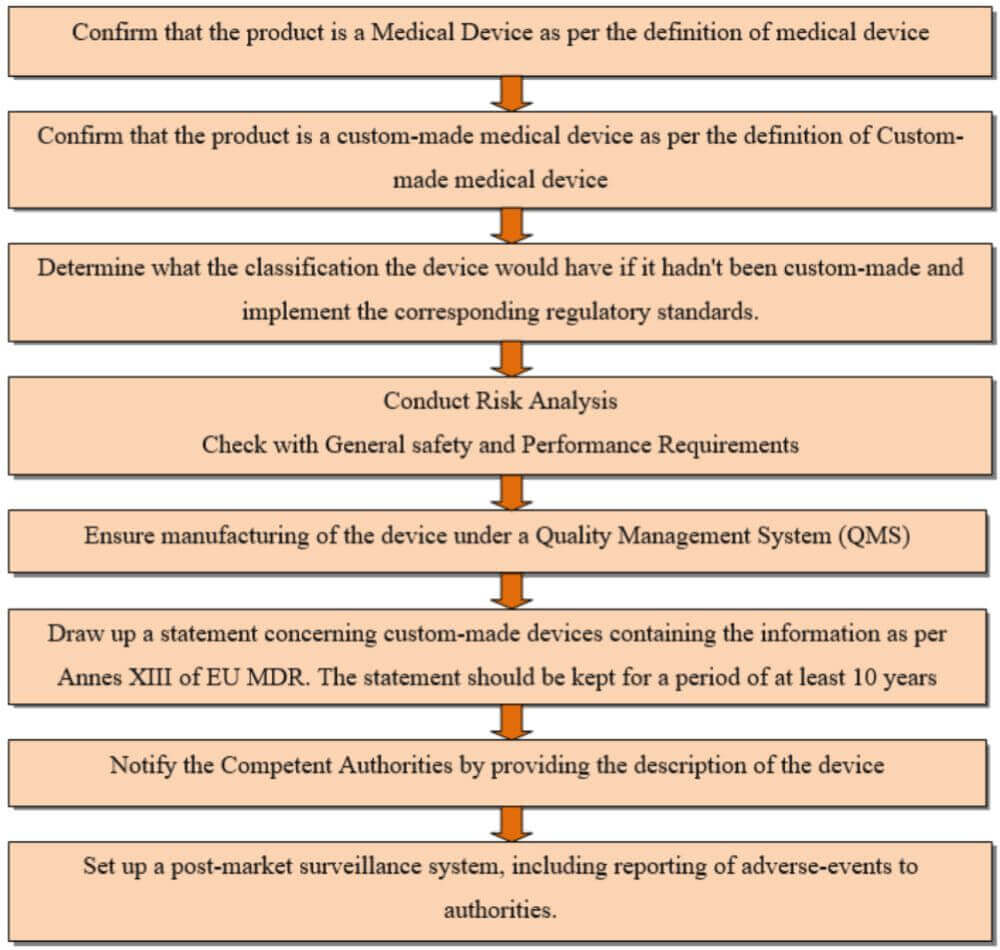
Procedure for custom made devices as per Annex XII of EU MDR
What is NOT needed?
Custom made medical devices are exempted from the requirements of the following –
Additionally, custom-made devices are not eligible for the CE mark, indicating that they have not undergone the standard conformity assessment procedures applicable to mass-produced devices.
*NOTE – There is no custom-made In-Vitro Diagnostic Devices permitted as per IVDR 2017/746
Additional specifics for class III custom-made implantable
High-risk custom-made medical devices require special attention and are a primary concern for the regulators. In order to address these risks, an additional requirement is imposed on Class III custom-made implantable devices, namely regarding conformity assessment.
In addition to adhering to the specifications detailed in Annex XIII, manufacturers must also undergo a conformity assessment as specified in Chapter I of Annex IX. The two main pathways for this assessment are either by adhering to the guidelines set forth in Annex IX, Chapter I, or by following Annex XI, Part A.
Furthermore, as per Annex XII of EU MDR, the statement referred to in the steps 6 of the ‘Procedure for custom made devices’ shall be retained for a period of at least 15 years
Conclusion
The goal of regulations pertaining to custom-made medical devices is to strike a balance between innovation and patient safety. By understanding and complying with these regulations, manufacturers can continue to develop innovative solutions tailored to individual patient needs, while healthcare professionals can confidently prescribe and utilize these devices to improve patient outcomes. Ultimately, a collaborative effort between regulators, manufacturers, healthcare professionals, and patients is essential to ensure the safe and effective use of custom-made medical devices in modern healthcare practice.
Reference
1. Medical Device Regulation (EU) 2017/745 of the European Parliament And Of The Council
2. IMDRF Final Document Personalized Medical Devices – Regulatory Pathways
3. MDCG 2021-3 Guideline: Questions and Answers on Custom-Made Devices & considerations on Adaptable medical devices and Patient-matched medical devices