Medical device manufacturing companies are facing more regulatory changes on a global basis than perhaps ever before. Most companies that distribute medical devices in Europe still have a lot to do before the new European Medical Device Regulation (MDR) takes effect in 2020.
The regulations have far-reaching impact, and they are why labelling has recently become a mission-critical business system for medical device companies.
Given that potentially thousands of labels will be impacted, now would be a good time to take the opportunity to rationalize the global labelling estate.
What Is Unique Device Identification (UDI)?
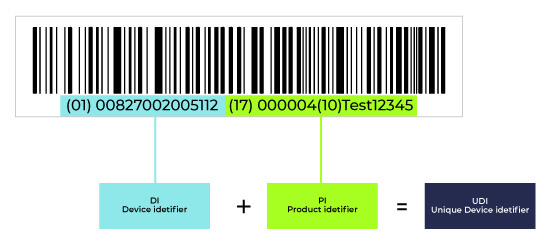
Unique Device Identification (UDI) is a system used to assign a unique identifier to medical devices, ensuring traceability and enhancing patient safety. The UDI consists of two key components:
Device Identifier (DI): A unique code specific to a device model or version, used to identify the manufacturer and device.
Production Identifier (PI): Includes details like batch/lot number, serial number, expiration date, or manufacturing date.
UDI enables better tracking, monitoring, and recall management of devices, and is mandated by regulatory authorities like the FDA and EU MDR.
UDI Requirements Under EU MDR
Scope:UDI applies to most medical devices and in vitro diagnostic devices (IVDs) under the EU MDR (Regulation (EU) 2017/745), including implantable, active, and reusable devices.
UDI Components:
Device Identifier (DI): Unique code identifying the specific model or version of a device.
Production Identifier (PI): Information such as batch/lot number, serial number, and expiration or manufacturing date.
Labeling: The UDI must be placed on the device, packaging, or both. It should be readable both in human-readable form and a machine-readable format (e.g., barcode, QR code).
EUDAMED Registration:Manufacturers must submit UDI data to the European Database on Medical Devices (EUDAMED) to facilitate device traceability and market surveillance.
Implementation Deadlines:
For high-risk devices (Class III and implantables), UDI compliance was required by May 26, 2021.
For lower-risk devices (Class IIa and IIb), the deadline is May 26, 2023.
Custom-made devices are exempt from UDI requirements.
Exceptions: Some devices, such as those for clinical investigations or certain low-risk devices, may be exempt from UDI requirements.
Advantages: UDI enhances patient safety, supports device traceability, improves post-market surveillance, and facilitates more efficient recalls if necessary.
UDI System Structure and Implementation
1.UDI Components:
Device Identifier (DI): A unique code assigned to each device model, identifying the manufacturer and device type.
Production Identifier (PI): Includes variable information such as batch/lot number, serial number, and expiration or manufacturing date.
2.UDI Format:
UDI must be presented in two forms:
Human-readable: Text format (e.g., alphanumeric code).
Machine-readable: Barcode, QR code, or RFID for easy scanning.
3.Global Databases:
UDI information is submitted to centralized databases like EUDAMED (EU) and GUDID (US) for regulatory tracking, post-market surveillance, and device traceability.
4.Device Labeling:
UDI must be included on the device label, packaging, or both, as per regulatory requirements.
5.Implementation Steps:
Manufacturers assign unique UDIs to devices, update labeling, and submit information to relevant regulatory bodies.
6.Benefits:
Improves traceability, patient safety, regulatory compliance, and device recall management.
Rationalizing Your Labelling Approach for EU MDR Compliance
By using the new regulations as an opportunity to cleanse their labelling data, device manufacturers can choose to make this the last time valuable resources are abstracted from day-to-day activities in order to hunt down the thousands of impacted labels.
The new EU MDR regulation puts labelling in a prominent position. It is critical for regulatory, labelling and operational professionals to gain a clear understanding of the impact of the new regulations on labelling.
For those not familiar with the acronym, an increasing number of regulatory bodies around the world, including the U.S. FDA and the European Commission, are requiring manufacturers to fulfill three primary requirements:
1)assign unique device identifiers (UDIs) to their products.
2)label those products with the UDIs in both human and machine (think bar code, RFID)
readable formats.
3)publish additional attributes about those products to regulatory databases.
UDIs consist of two numbers: A Device Identifier (DI) relating to the specific device model and its manufacturer, and an optional Production Identifier (PI) that provides such details as lot or batch number, serial number, manufacture date, expiration date, and any other important production-related data. All mandatory information must be included both as human-readable plain text and in a format suitable for automatic identification and capture (ASIC).
There is no simple shortcut to this part of the process, but with the right approach, it becomes a downstream value-creating activity rather than a short-term business overhead.
And here is where Maven can help you.
Maven Profcon Services is a Medical Device Consultant, provider of regulatory solutions for all size of industries not only India, but globally.
Act Now: Streamline Labeling for EU MDR Compliance
It’s important to remember, however, that no software solution is going to deliver a benefit without first carrying out the rationalization and consolidation activities discussed above. Organizations adopting this approach will find themselves better equipped to respond to future legislative changes, new market opportunities, and the inevitable day-to-day labeling challenges in bringing new products to market.
Benefits of UDI Compliance for Manufacturers
1.Enhanced Traceability: UDI enables better tracking and tracing of devices throughout the supply chain, ensuring quick identification and response in case of recalls or safety concerns.
2.Improved Regulatory Compliance: Meeting UDI requirements ensures manufacturers adhere to global regulatory standards (e.g., EU MDR, FDA), avoiding penalties and facilitating smoother market access.
3.Efficient Post-Market Surveillance: UDI supports continuous monitoring of device performance and safety through databases like EUDAMED and GUDID, helping manufacturers maintain a proactive approach to product quality.
4.Streamlined Inventory Management: UDI enables more accurate inventory control, reducing errors, optimizing stock levels, and improving the efficiency of logistics and distribution.
5.Facilitated Device Recall Management: With UDI, manufacturers can quickly identify affected devices, improving recall efficiency and minimizing patient risk.
6.Harmonization: UDI provides manufacturers with a standardized identification system, easing market entry into multiple regions (e.g., EU, US, and other countries with UDI regulations).
7.Reduced Costs: By streamlining supply chain processes, improving inventory management, and reducing labeling errors, UDI compliance can lead to long-term cost savings for manufacturers.
8.Enhanced Patient Safety: UDI helps ensure accurate device identification, which is critical for patient safety, particularly in cases of device recalls or adverse events.
9.Competitive Advantage: Manufacturers who implement UDI early may gain a competitive edge by demonstrating their commitment to safety, regulatory compliance, and operational efficiency.
In the end, UDI compliance is a fact of life for manufacturers. They must either comply with the rule or risk no longer being able to sell their products in a given market. How a manufacturer approaches UDI compliance is less certain and can make the difference between more regulatory burden and higher costs of doing business or the opportunity to build better relationships with customers and building better products for patient care.
Decide What’s Best for Your Organization
1.Understand Regulatory Obligations: Ensure awareness of UDI requirements under EU MDR, FDA, and other regional regulations. 2.Assess Device Portfolio: Review device types and classifications to prioritize UDI compliance for high-risk devices. 3.Establish Internal Processes: Develop procedures for assigning UDIs, data entry, and label management. 4.Labeling Technology: Invest in technology for both human-readable and machine-readable UDI formats (barcodes, QR codes). 5.Staff Training: Educate employees on UDI standards, processes, and data management. 6.Post-Market Surveillance: Set up systems for ongoing monitoring and database updates. 7.Evaluate Costs: Analyze the cost of UDI implementation versus long-term benefits in safety and compliance.