Hello Readers!
A medical device manufacturer has to follow Conformity Assessment Procedure before placing the device on the EU Market. These conformity assessment procedures are documented in Article 52 of the EU MDR 2017/745 for Medical Devices & Article 48 of EU IVDR 2017/746 for Invitro Diagnostic Medical Device. There are number routes listed in this Articles, the manufacturer has to select the route depending on the Class and Type of the Device.
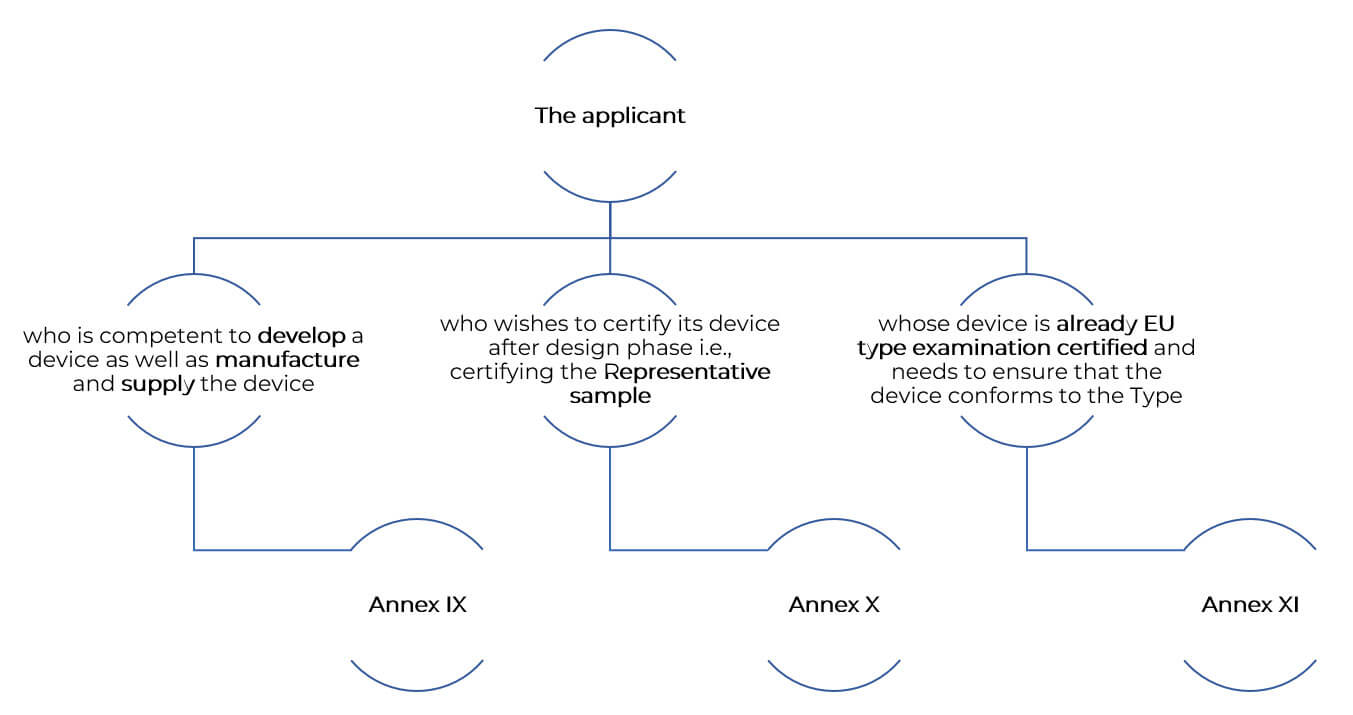
Conformity assessment is depended on following key annexures of the regulations. Those are:
Annex IX- CONFORMITY ASSESSMENT BASED ON A QUALITY MANAGEMENT SYSTEM AND ON ASSESSMENT OF TECHNICAL DOCUMENTATION.
Annex X- CONFORMITY ASSESSMENT BASED ON TYPE-EXAMINATION.
Annex XI- CONFORMITY ASSESSMENT BASED ON PRODUCT CONFORMITY VERIFICATION.
Let us understand the concept of these annexures in detail.
ASSESSMENT BASED ON A QUALITY MANAGEMENT SYSTEM AND ON ASSESSMENT OF TECHNICAL DOCUMENTATION
- The manufacturer is supposed to maintain a Quality Management System and it shall be subjected to Audit and Surveillance.
- The notified body will audit the Quality Management System to verify its compliance against the regulatory requirements, this audit will be conducted at manufacturer’s premises or on the premises of the manufacturer’s suppliers
and/or subcontractors as per the requirement. After Audit, if the requirements are met, the notified body shall issue EU quality management system certificate. - For Class IIa, Class IIb & Class III medical device & Class C & Class D IVD Devices, surveillance audit shall be conducted at least once in a year and an unannounced audit at least once in 5 years. During the unannounced audit the
Notified Body shall test adequate sample of the device from the Market or from the manufacturing Process and if any non-compliance is found they shall suspend or withdraw or add Restrictions on the Certificate. - During the assessment of Technical Documentation as per Annex II & Annex III, the notified body shall also conduct required test in relation to the device or may ask the manufacturer to carry those tests. After the assessment, the
Notified body will issue the EU technical documentation assessment certificate. - When any substantial change is planned by the manufacturer, it is supposed to be informed to the notified body. The notified body will assess the change and conduct audit if necessary and a supplement to the EU QMS certificate & EU
Technical documentation assessment certificate will occur.
ASSESSMENT BASED ON TYPE-EXAMINATION
- In this approach the notified body certifies the device on the basis of its representative sample production envisaged, its technical documents and its life cycle processes. To be clear Production envisaged means the device has not
yet arrived at production phase, it shall be produced in future on the basis of this representative sample once it is EU Certified. - The notified body shall be kept available with the representative sample by the manufacturer/applicant. As per regulatory requirement, relevant device tests shall be carried out by the Notified body or they may ask the applicant to
provide those test results. The Examination and Assessment of Technical documentation shall be performed by the Notified Body and they shall map out a report. EU type Examination Certificate shall be issued. - Approval from the Notified Body is required before changes (which may affect the GSPR or with the conditions prescribed for use) to the approved device is planned. A supplement to the EU Type Examination Certificate shall be issued
after examination of the planned change by the Notified Body.
ASSESSMENT BASED ON PRODUCT CONFORMITY VERIFICATION
- This approach is used to ensure that the device which is already issued an EU Type Examination certificate conforms to the TYPE and meet the regulatory requirement.
- There are two possible approaches i.e., Production Quality Assurance and Product Verification.
- In Production Quality Assurance, the manufacturer shall implement and maintain the Quality management system and shall follow the surveillance activity. The notified body shall issue an EU Quality assurance Certificate if the device
conforms to the Type described in EU Type examination certificate. - In Product Verification, after examination of each manufactured device the manufacturer shall issue EU Declaration of Conformity. Examination and testing of Each Product shall be carried out by the Notified body as per the
regulatory requirements. The Notified body shall fix or have fixed its identification number on each approved device and shall write down an EU product verification certificate.
The Query – Which approach shall be selected depending on the applicants Requirements?
Following image illustrates all the routes with respect to the class of the devices as per EU MDR 2017/745 and EU IVDR 2017/746 respectively.
Conformity Assessment Routes
Class I-MDR
Class I | Class Is/Im/Ir |
| |
| Annex II & III Technical Documentation | | Annex II & III Technical Documentation | Annex II & III Technical Documentation | |
| Declaration of Conformity (Annex IV) | | Annex IX QMS Chapter I, III | Annex XI – PART A Production Quality Assurance | |
| CE marking (Annex V) | | Declaration of Conformity (Annex IV) | Declaration of Conformity (Annex IV) | |
| Note: No Notify Body Involvement | | CE marking (Annex V) | CE marking (Annex V) | |
Class II-MDR
Class II a | Class IIb (Annex VIII Rule 12 devices) | Class II b implantable WET,
Class II b non implantable, non Rule 12, non WET | Class II b implanatble devices (excluding WET) |
| | | | |
| Annex IX QMS Chapter I, III | Annex XI – PART A (Production Quality Assurance) | Annex XI – PART B (Production Verification) | | | Annex IX QMS Chapter I, III | Annex X Type Examination | Annex X Type Examination | | | Annex IX QMS Chapter I, III | Annex X Type Examination | Annex X Type Examination | | | Annex IX QMS Chapter I, III | Annex X Type Examination | Annex X Type Examination | |
| Annex IX Chapter II Technical Documentation (Assessed per device category) | Annex II & III Technical Documentation (assesed per device category) | Annex II & III Technical Documentation (assesed per device category) | | | Annex IX Chapter II Technical Documentation (Assessed per generic device group) | Annex XI – PART A (Production Quality Assurance) | Annex XI – PART B (Production Verification) | | | Annex IX Chapter II Technical Documentation (Assessed per generic device group) | Annex XI – PART A (Production Quality Assurance) | Annex XI – PART B (Production Verification) | | | Annex IX Chapter II Technical Documentation (Technical documenatation for every device) | Annex XI – PART A (Production Quality Assurance) | Annex XI – PART B (Production Verification) | |
| Declaration of Conformity (Annex IV) | Declaration of Conformity (Annex IV) | Declaration of Conformity (Annex IV) | | | Clinical Evaluation Consultation Procedure Annex IX Sec 5/ Annex X Sec 6 | Clinical Evaluation Consultation Procedure Annex IX Sec 5 / Annex X Sec 6 | Clinical Evaluation Consultation Procedure Annex IX Sec 5 / Annex X Sec 6 | | | Declaration of Conformity (Annex IV) | Annex XI – PART B (Production Verification) | Declaration of Conformity (Annex IV) | | | Declaration of Conformity (Annex IV) | Declaration of Conformity (Annex IV) | Declaration of Conformity (Annex IV) | |
| CE marking (Annex V) | CE marking (Annex V) | CE marking (Annex V) | | | Declaration of Conformity (Annex IV) | Declaration of Conformity (Annex IV) | Declaration of Conformity (Annex IV) | | | CE marking (Annex V) | CE marking (Annex V) | CE marking (Annex V) | | | CE marking (Annex V) | CE marking (Annex V) | CE marking (Annex V) | |
| | | CE marking (Annex V) | CE marking (Annex V) | CE marking (Annex V) | | | |
Class III-MDR
Class III Implantable devices (Including devices containing medicinal substance, Human tissue or animal tissue) | Class III non-Implantable devices (Including devices containing medicinal substance, Human tissue or animal tissue) | Custom-made Class III Implantable devices | Custom-made devices (Excluding Custom-made Class III Implantable devices) |
| | | | |
| Annex IX QMS Chapter I, III | Annex X Type Examination | Annex X Type Examination | | | Annex IX QMS Chapter I, III | Annex X Type Examination | Annex X Type Examination | | | Annex XIII Documentation | Annex XIII Documentation | | |
| Annex IX Chapter II Technical Documentation (Technical documenatation for every device) | Annex XI – PART A (Production Quality Assurance) | Annex XI – PART B (Production Verification) | | | Annex IX Chapter II Technical Documentation (Technical documenatation for every device) | Annex XI – PART A (Production Quality Assurance) | Annex XI – PART B (Production Verification) | | | Annex IX QMS Chapter I | Annex XI – PART A (Production Quality Assurance) | | | Collect PMS, PMCF data as per Part B of Annex XIV | |
| Consultation 2001/83/EC, EC/726/2004, 2004/23/EC, EU/722/2012 | Consultation 2001/83/EC, EC/726/2004, 2004/23/EC, EU/722/2012 | Consultation 2001/83/EC, EC/726/2004, 2004/23/EC, EU/722/2012 | | | Consultation 2001/83/EC, EC/726/2004, 2004/23/EC, EU/722/2012 | Consultation 2001/83/EC, EC/726/2004, 2004/23/EC, EU/722/2012 | Consultation 2001/83/EC, EC/726/2004, 2004/23/EC, EU/722/2012 | | | Statement Annex XIII Section 1 | Statement Annex XIII Section 1 | | | Statement Annex XIII Section 1 | |
| Clinical Evaluation Consultation Procedure Annex IX Sec 5/ Annex X Sec 6 | Clinical Evaluation Consultation Procedure Annex IX Sec 5/ Annex X Sec 6 | Clinical Evaluation Consultation Procedure Annex IX Sec 5/ Annex X Sec 6 | | | Declaration of Conformity (Annex IV) | Declaration of Conformity (Annex IV) | Declaration of Conformity (Annex IV) | | | CE Certificate issued | CE Certificate issued | | | Note: No Notify Body Involvement | |
| Declaration of Conformity (Annex IV) | Declaration of Conformity (Annex IV) | Declaration of Conformity (Annex IV) | | | CE marking (Annex V) | CE marking (Annex V) | CE marking (Annex V) | | | |
| CE marking (Annex V) | CE marking (Annex V) | CE marking (Annex V) | | | | |
Class A- IVDR
Class A (Excluding Sterile Device) | Class A (Sterile Device) |
| |
| Annex II and III Technical Documentation | | Annex II and III Technical Documentation | Annex II and III Technical Documentation | |
| Declaration of conformity (Annex IV) Self declaration | | Annex IX* QMS Chapters I, III | Annex XI – PART A Production Quality Assurance | |
| CE marking (Annex V) | | Declaration of conformity (Annex IV) | Declaration of conformity (Annex IV) | |
| Note: No Notify Body Involvement | | CE Marking (Annex V) | CE Marking (Annex V) | |
Class B-IVDR
Class B (Excluding self testing & Near Patient Testing Devices) | Class B (Self Testing and NPT Devices) |
| Annex IX QMS Chapters I, III | Annex IX QMS Chapters I, III |
| Annex IX Chapter II Technical Documentation assessed per device category (Section 4) | Annex IX Chapter II Technical Documentation assessed for every device (Sections 4 and 5.1) |
| Declaration of conformity (Annex IV) | Declaration of conformity (Annex IV) |
| CE Marking (Annex V) | CE Marking (Annex V) |
Class C- IVDR
Class C (Excluding Self testing, NPT and companion Diagnostic) | Class C (Self Testing and near patient Tetsting) | Class C (Companion Diagnostic) |
| Annex IX QMS Chapters I, III | Annex IX QMS Chapters I, III | Annex IX QMS Chapters I, III |
| Annex IX Chapter II Technical Documentation assessed per generic device group (Section 4) | Annex IX Chapter II Technical Documentation assessed for every device (Sections 4 and 5.1) | Annex IX Chapter II Technical Documentation assessed for every device (Sections 4 and 5.2) |
| Declaration of conformity (Annex IV) | Declaration of conformity (Annex IV) | CA or EMA consultation as per Annex IX – Section 5.2 |
| CE Marking (Annex V) | CE Marking (Annex V) | Declaration of conformity (Annex IV) |
| | CE Marking (Annex V) |
Class D-IVDR
Class D
(with common specification)- excluding CDx | Class D
(with no common specification)- excluding CDx | Class D
CDx Devices |
| Annex IX QMS Chapters I, III | Annex IX QMS Chapters I, III | Annex IX QMS Chapters I, III |
| Annex IX Chapter II excluding Section 5 Technical Documentation assessed for every device (For self-test and NPT Section 5.1 is included) | Annex IX Chapter II excluding Section 5 Technical Documentation assessed for every device (For self-test and NPT Section 5.1 is included) | Annex IX Chapter II including Section 5.2 Technical Documentation assessed for every device |
| Verification by EU Reference Laboratory Annex IX – Section 4.9 (Required for devices for which one or more EU Reference Laboratories have been designated in accordance with Article 100) | Verification by EU Reference Laboratory Annex IX – Section 4.9 (Required for devices for which one or more EU Reference Laboratories have been designated in accordance with Article 100) | CA or EMA consultation – Annex IX, Section 5.2 |
| Declaration of conformity (Annex IV) | Expert consultation (Article 48(6))(Where no Common Specification is available, the NB shall provide the performance evaluation report of the manufacturer to the Expert Panel within five days of receipt. Required in cases where the device is the first of its type) | Verification by EU Reference Laboratory Annex IX – Section 4.9(Required for devices for which one or more EU Reference Laboratories have been designated in accordance with Article 100) |
| CE Marking (Annex V) | Declaration of conformity (Annex IV) | Expert consultation (Article 48(6)) (Where no Common Specification is available, the NB shall provide the performance evaluation report of the manufacturer to the Expert Panel within five days of receipt. Required in cases where the device is the first of its type) |
| CE Marking (Annex V) | Declaration of conformity |
| | CE Marking (Annex V) |