Introduction to ISO 14971 and Risk Management in Medical Devices
ISO 14971 is an internationally recognized standard specifically developed for the application of risk management to medical devices. It provides a systematic framework for identifying hazards, estimating and evaluating associated risks, implementing control measures, and monitoring the effectiveness of these controls throughout the product lifecycle. The primary goal of ISO 14971 is to ensure patient safety and device effectiveness by minimizing potential harm that could arise from device use, misuse, or malfunction.
Risk management is a critical component of medical device design, development, manufacturing, and post-market surveillance. By adopting ISO 14971, manufacturers demonstrate their commitment to compliance and product safety, aligning with global regulatory expectations such as those from the EU MDR and US FDA. The standard emphasizes the importance of proactive risk identification, evidence-based decision-making, and maintaining comprehensive documentation in a risk management file. With ISO 14971, organizations can ensure that their devices are not only safe but also reliable and trustworthy in clinical settings.
Objectives and Importance of Risk Management
The primary objective of risk management in the medical device industry is to ensure patient safety, device effectiveness, and regulatory compliance throughout the product lifecycle. A robust risk management process helps identify potential hazards early in the development phase, thereby reducing the chances of product recalls, regulatory penalties, and harm to users. It also aids in decision-making by providing a structured approach to evaluate and mitigate risks systematically.
Risk management is not only a regulatory requirement but also a strategic tool that improves product design and performance. It ensures that all identified hazards are controlled within acceptable limits, thereby enhancing user confidence and supporting market acceptance. Moreover, ongoing risk evaluation during post-production helps identify any emerging risks that may arise due to field usage, thereby promoting continuous improvement.
Overall, risk management as per ISO 14971 plays a vital role in demonstrating a manufacturer’s commitment to safety, quality, and compliance—factors that are critical for long-term success in the medical device industry.
Roles and Responsibilities in Risk Management
In an effective risk management system as per ISO 14971, clearly defined roles and responsibilities are essential to ensure consistency, traceability, and accountability throughout the lifecycle of a medical device.
Top management is responsible for establishing a risk management policy, defining risk acceptability criteria, and ensuring the availability of adequate resources. They must also demonstrate leadership and commitment to maintaining an effective risk management process.
Risk management team members—including design engineers, quality assurance personnel, regulatory affairs specialists, and clinical experts—are responsible for identifying hazards, estimating and evaluating risks, and implementing appropriate risk control measures. These individuals must be competent and trained in the application of ISO 14971 and the intended use of the medical device.
Regulatory and quality professionals ensure that documentation aligns with applicable standards and regulatory expectations. They are responsible for maintaining the risk management file, reviewing post-market surveillance data, and supporting audits.
Manufacturing and service teams are also involved in monitoring production and post-production data, contributing valuable information for ongoing risk evaluation and residual risk management.
The process flow for risk management based on ISO 14971 is as below According to clause 3 in ISO 14971, top management must:
- Exhibit commitment for managing risks of each medical device.
- Establish a policy and the acceptance criteria for a risk.
- Review the risks for medical devices at planned intervals (reviews can be performed at management reviews).
As with other management standards, people who perform risk assessment should be competent and knowledgeable (e.g., through trainings & experience on ISO 14971, medical device application, etc.).
Suggested Read: Risk Management for Medical Devices
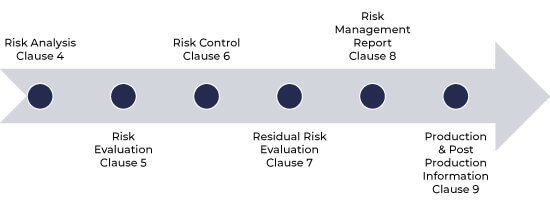
Risk management process flow as per ISO 14971
Risk management file
Another important element in risk management (to ensure traceability) is a risk management file, which is established for every medical device. The file is used to keep record of:
- Risk analysis results.
- Risk evaluation results.
- Risk control measures.
- Residual risk evaluation results for each identified hazard.
The risk management file will be used to gather all information related to risk, even in post-production situations.
Risk Management Process as per ISO 14971
The process of risk management has the following steps:
Risk Analysis Techniques
Risk analysis is performed on each medical device, and possible hazards are identified. Risk is estimated for each hazardous situation. Characteristics that can foreseeably affect the safety of the medical device are also listed. Risk analysis should also incorporate a combination of hazardous events that can result in a hazardous situation, whereas reasonably foreseeable combinations of such events should be analyzed separately. For example, when a heel stick is used to collect blood from infants for testing, the blood is warmed with a chemical pack. The sudden rupturing of this chemical pack is a foreseeable effect of the characteristics of the chemical pack, and the hazardous event is a combination of the heel stick used for collecting the sample (likely a negligible hazard) and the chemical pad used to ease the process of sampling. The risk management file is updated accordingly based on all analysis results.
Suggested Read: Risk Analysis: Understanding the Quantification of Benefit
Risk Evaluation Criteria
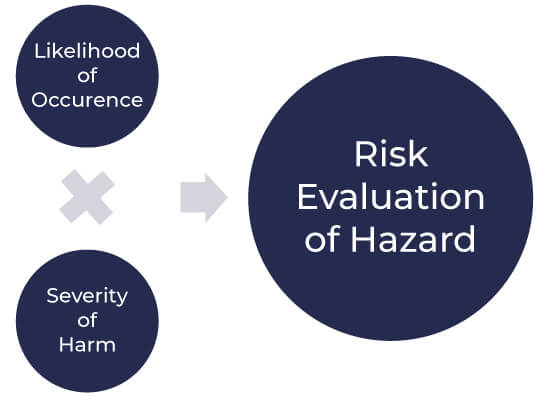
Each hazardous situation is studied, and then the organization’s risk acceptability criteria are used to confirm whether risk reduction is needed for this hazard or not. The results of risk evaluation activities are also recorded in the file. Risk evaluation is normally done by multiplying the severity of the hazard by the likelihood of its occurrence.
Risk evaluation as per ISO 14971
Risk Control Strategies
Risk control is a risk reduction process in which an unacceptable risk is minimized. The effectiveness of the control is measured by reevaluation of residual risk, i.e., remaining risk after the control is implemented. Sometimes, controls allocated to minimize a risk add another risk hazard – such controls are ineffective until, and unless, the new risks are within acceptable range or controlled within acceptable limits. A risk control is chosen from the available options based on the following factors:
- Practicality (how useful the implemented control is).
- Simplicity (how easily it can be implemented).
- Economic feasibility (the cost of the control does not affect product profitability).
When implemented, risk controls are verified. If the residual risk is unacceptable, a risk benefit analysis is conducted. If an additional control is impractical, then the risk benefit analysis should dictate whether the medical benefits of the device outweigh the residual risk. Records of each step of risk control are maintained in the risk management file, which includes control options, selection of control, risk control review, control verification, residual risk calculation, risk benefit analysis, etc.
Residual Risk and Benefit-Risk Analysis
Residual risk evaluation is done after all controls are in place and effective. A file is maintained with the risk management register after all risks have been properly controlled, and records are maintained. Any change may require reevaluation of overall residual risks.
Risk management report
Just as management reviews are planned for the Quality Management System, likewise, such reviews should be planned for the risk management system. Before a medical device enters the commercial market, a review should be conducted. Based on the review, a risk management report is prepared. The report should include the results of the review and be incorporated into the risk management file as mentioned ISO 14971.
Information from production and post-production
A system for monitoring the performance of the medical device should be developed, established, and maintained as mentioned in ISO 14971. The results should be recorded in the risk management file. Information that comes from production includes any defects or failures in clinical trials, and results of post-production include any customer complaints or product failures that may increase the risk (because of increased likelihood of occurrence).
Suggested Read: Active Medical Devices: Effective Risk Management Strategies
Management commitment to control risk of a medical device as per ISO 14971
With the help of a risk management system based on ISO 13485 and ISO 14971, each phase of a risk management cycle is documented comprehensively to demonstrate the manufacturer’s commitment to controlling risk in the life of the medical device. A strong risk management system as per ISO 14971 also provides significant value by helping with the development, manufacture, and delivery of new medical devices. Devices under development are subject to higher levels of scrutiny. Also, ISO 14971 risk management system helps with documenting modifications to ensure product safety, functionality, and usability.
At Maven, we have a team of Experts in product knowledge as well as Standard compliance. We can tailor your Risk management report as per ISO 14971 according to your product requirements.