Notified Body means a conformity assessment body designated in accordance with this Regulation.
The role of a Notified Body is to conduct a conformity assessment under the relevant EU Directives. The Notified Body conducts the conformity assessment against the relevant sections of the applicable Directive (MDD, AIMDD or IVDD). The conformity assessment usually involves an audit of the manufacturer’s quality system and depending upon the particular classification of the device, a review of the relevant technical documentation provided by the manufacturer in support of the safety and performance claims for the device.
II. Expert Panels
The Need:
Although the Commission has considerable in-house expertise, it needs specialist advice from outside experts as a basis for sound policymaking. This may be provided by groups of experts or external consultants, or take the form of studies.
A consultative body:
Set up by the Commission or its departments to provide them with advice and expertise composed of public and/or private sector members which meets more than once.
Gathering expertise from various sources may include gathering the views of various stakeholders.
There are 2 types of Commission expert groups:
Formal- set up by Commission decision.
Informal- set up by an individual Commission department that has obtained the agreement of the Commissioner and Vice-President responsible and of the Secretariat-General.
Responsibility:
They advise the Commission in relation to:
the preparation of legislative proposals and policy initiatives.
the preparation of delegated acts.
the implementation of EU legislation, programs and policies, including coordination and cooperation with Member States and stakeholders in that regard.
Where necessary, the preparation of implementing acts at an early stage, before they are submitted to the committee in accordance with Regulation (EU) No 182/2011.
Expert groups are not set up to engage in general debate with stakeholders or the public; rather, they provide a forum for discussion on a given subject and on the basis of a specific mandate involving high-level input from a wide range of sources and stakeholders that takes the form of opinions, recommendations and reports.
This input is not binding on the Commission, which remains fully independent regarding the way it takes into account the expertise and views gathered. When proposing new policies and measures, the. Commission always tries to find the best solution in the general interest of the EU and its Member States.
Moreover, experts groups are not the Commission’s only source of expert input. When gathering the full range of views on an issue, it also has recourse to studies, European agencies, green papers, public consultations, hearings, and so on. Overall stakeholder participation and representation should therefore always be seen in the light of all the initiatives the Commission takes.
III. Medical Device Coordination Group (MDCG)
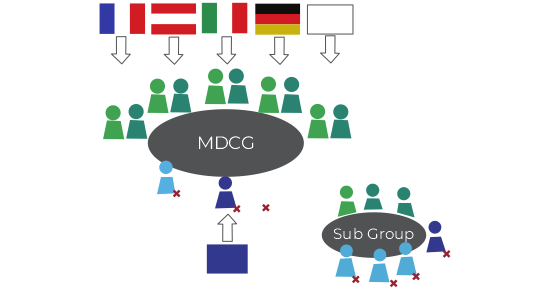
Article 103 of the MDR and Article 98 of the IVDR require the European Commission (Commission) to establish a corresponding Medical Device Coordination Group (MDCG). The MDCG, characterized as a centralized organization of qualified individuals with medical device and in vitro device experience, will function at the request of the commission to develop common specifications, draft guidance documents, oversee and approve applications by conformity assessment bodies, and resolve regulatory or other product lifecycle issues. The MDCG will work cooperatively to ensure consistent cross-border decisions and enforcement. Each of the 27 member states must appoint one individual expert and one alternate expert to represent the competent authority of their member state and serve a renewable three-year term on each MDCG. If qualified, an individual may serve in the same or alternate capacity on both MDCGs. A non-voting representative of the commission will chair each MDCG. The MDCG is required to meet at regular intervals and at the request of the commission or a member state. Figure 1 illustrates the membership structure of the MDCG.
Responsibility:
Assessment of applicant conformity assessment bodies.
Advise commission in matters concerning the coordination group of notified body.
Guidance development to ensure regulation harmonization.
Guidance development for designating and monitoring notified body
Guidance development for application of general safety and performance requirement.
Guidance development for conduct of clinical evaluations and investigation.
Guidance development for vigilance activities.
Monitoring of technical progress and assessing adequacy of.
general safety and performance requirements of regulation.
Contribute to development of device and common specification standards, scientific guidelines and clinical investigation guidelines for implantable devices and Class III devices.
Assist member state competent authorities in classification and regulatory status determinations, clinical investigation and market surveillance.
Work with commission to set up a unique device identification database.
Work with commission to set up an electronic system for.
The European Commission is the EU’s politically independent executive arm. It is alone responsible for drawing up proposals for new European legislation, and it implements the decisions of the European Parliament and the Council of the EU.
Responsibility:
Proposes new laws
The Commission is the sole EU institution tabling laws for adoption by the Parliament and the Council that:
Protect the interests of the EU and its citizens on issues that can’t be dealt with effectively at national level; get technical details right by consulting experts and the public
Manages EU policies & allocates EU funding
Sets EU spending priorities, together with the Council and Parliament.
Draws up annual budgets for approval by the Parliament and Council.
Supervises how the money is spent, under scrutiny by the Court of Auditors
Enforces EU law
Together with the Court of Justice, ensures that EU law is properly applied in all the member countries.
Represents the EU internationally
Speaks on behalf of all EU countries in international bodies, in particular in areas of trade policy and humanitarian aid.
Negotiates international agreements for the EU
V. Person Responsible for Regulatory Compliance
The person in charge with regulatory compliance assumes a role of greater responsibility. In particular, their responsibilities include.
Checking the compliance of the devices with the Quality Management System before they are released.
Verifying that the technical documentation and the EU declaration of conformity are drawn up and updated.
Managing the post-market surveillance obligations and ensuring that they are met.
Managing the reporting obligations and ensuring that they are met.
Qualifications of the Person Responsible for Regulatory Compliance.
The EU MDR and IVDR stipulate that the PRRC must have:
Requisite expertise in the field of medical devices, and
A university degree, diploma, or some other formal qualifications, and
At least one year of experience in regulatory affairs or quality systems management related to medical devices.
If no university degree, diploma, or other qualification, must have four years of professional experience in regulatory affairs or quality systems management related to medical devices.