The MDD to MDR transition process involves updating medical device compliance from the Medical Device Directive (MDD) to the Medical Device Regulation (MDR). This shift includes stricter classification rules, enhanced clinical evaluation, and increased post-market surveillance. Companies must conduct a gap analysis to ensure compliance with MDD and MDR requirements.
Step #1 Verify your product’s classification
The first step in the MDD to MDR transition is verifying your product’s classification under EU MDR Annex VIII. The MDD vs MDR classification rules have changed, with MDR introducing a stricter, risk-based approach. Some devices, including software and reusable surgical instruments, now fall into higher-risk categories. Proper classification ensures compliance with the correct conformity assessment route. Manufacturers should review the new rules and consult their Notified Body if needed. Misclassification can lead to delays in certification, affecting market access. Conducting a thorough classification review is essential for a smooth MDD to MDR transition.
Step #2 Update the documentation and labelling
As part of the MDD to MDR transition, updating documentation and labeling is crucial. The MDR introduces stricter requirements, demanding more detailed technical files, risk assessments, and clinical evaluations. Manufacturers must ensure that labels meet the new UDI requirements and provide clear, updated information in compliance with MDR standards. Conducting an MDD vs MDR gap analysis helps identify necessary changes in labeling, ensuring accuracy and regulatory alignment. Understanding MDD and MDR differences is essential to avoid compliance risks. Proper documentation updates streamline the certification process and facilitate a smooth transition to MDR, ensuring continued market access.
Step #3 Make sure your product meets the UDI requirements
A critical aspect of the MDD to MDR changes is compliance with Unique Device Identification (UDI) requirements. Unlike MDD, MDR mandates UDI for all medical devices, enhancing traceability and market surveillance. Manufacturers must assign a UDI to each product, ensuring it aligns with MDR standards and is registered in EUDAMED. Proper UDI implementation helps streamline post-market monitoring and regulatory compliance. Understanding the differences between MDD and MDR regarding UDI is essential for a smooth transition. Conducting a gap analysis will help identify necessary updates and ensure your product is fully compliant with MDR’s stricter identification rules.
Step #4 Choose a Notified Body
Selecting a Notified Body is a crucial step in the MDD to MDR transition process. Under the difference between MDD and MDR regulations, MDR has stricter requirements, and fewer Notified Bodies are designated under the new framework. Manufacturers must ensure their chosen Notified Body is accredited for their device classification. Early engagement is essential due to high demand and longer review timelines. Understanding EU MDR and MDD differences will help streamline the certification process and avoid delays. Companies should verify the scope of their Notified Body’s designation and prepare comprehensive technical documentation to meet MDR compliance.
Step #5 Ensure you’re meeting all the obligations of a manufacturer
To comply with EU MDR, manufacturers must fulfill all regulatory obligations, including robust clinical evaluation, risk management, and post-market surveillance. Ensuring technical documentation meets MDR standards is crucial for maintaining certification. Unique Device Identification (UDI) and active post-market clinical follow-up (PMCF) are mandatory under the new regulation. Manufacturers should also align their quality management system (QMS) with ISO 13485 and MDR requirements. Conducting a thorough gap analysis helps identify compliance gaps early, preventing delays in certification. Staying updated with evolving Notified Body expectations ensures a smooth transition from MDD to MDR without regulatory setbacks.
Step #6 Get audited and certified
Once your documentation aligns with MDR requirements, the next crucial step is undergoing an audit by a Notified Body. This audit verifies compliance with MDR, focusing on technical documentation, clinical evaluation reports, and post-market surveillance plans. Passing the audit is essential for obtaining MDR certification, allowing continued market access in the EU. Ensure your quality management system meets ISO 13485 standards and that all risk management processes align with MDR guidelines. Proactive planning and expert guidance can streamline the certification process, reducing delays and ensuring regulatory approval for your medical devices.
Step #7 Continuous compliance
Achieving MDR certification is not a one-time process—it requires continuous compliance to maintain market access. Medical device manufacturers must establish robust post-market surveillance (PMS) systems, regularly update clinical evaluation reports (CERs), and conduct risk management reviews. Implementing a quality management system (QMS) aligned with ISO 13485 ensures ongoing adherence to MDR requirements. Regular audits, Periodic Safety Update Reports (PSURs), and proactive engagement with Notified Bodies help identify and mitigate compliance risks. Staying updated on EU MDR regulatory changes is crucial for long-term success and smooth market operations.
Europe’s new Medical Devices Regulation(MDR) will bring significant regulatory changes that may impact multiple business units within your organization.
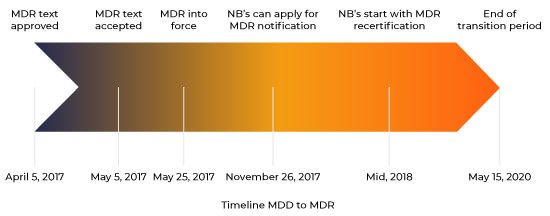
On April 5th 2017, the new Medical Device Regulation (2017/745/EU) was accepted by the European parliament. On May 5th 2017 it was published in the European Official Journal (EUOJ), based on which it came into force 20 days later. As a consequence of this, a series of events was initiated which will cause a lot of changes for the producers as well as for the Notified Bodies.
MDD to MDR Transition Timeline
First of all, the directive is replaced by a regulation. Different from a European directive, a regulation has a direct effect. This means that the regulation does not have to transfer into a national legislation.
Unclear Classification Under EU MDR Annex VIII: What Are the Options?
Conformity assessments for many medical devices have changed under the MDR, and classification must be reevaluated based on the new rules. The changes in the conformity route could require additional performance testing and/or clinical evaluation. Evaluating your device classification is essential to ensure that your company can allocate the time necessary to keep marketing their products in the EU. However, some classifications will remain unclear for some manufacturers, and companies will need to contact their Notified Body for assistance to make sure they understand the proper classification and conformity route. In some cases, if there is a dispute, the classification request will be forwarded to the competent authority for a final decision. If your device classification has changed, you should be able to pursue MDD certification renewal and that may be advantageous if you are unsure whether your device has adequate clinical data to support MDR certification.
Key Differences Between MDD and MDR
The terms MDD (Medical Device Directive) and MDR (Medical Device Regulation) refer to frameworks governing medical devices in the European Union (EU). MDR replaced MDD to address shortcomings and enhance safety, transparency, and market surveillance. Here’s a comparison of their key differences:
1. Regulatory Status
MDD (93/42/EEC): A directive that required transposition into national laws, leading to variations in implementation across EU member states.
MDR (2017/745): A regulation directly applicable across all EU member states, ensuring uniform implementation.
2. Scope
MDD: Focused primarily on traditional medical devices and active implantable devices.
MDR: Expanded scope to include products without an intended medical purpose (e.g., cosmetic contact lenses, aesthetic fillers) and devices for cleaning, sterilizing, or assisting other devices.
3. Classification Rules
MDD: Relied on a set of classification rules with less emphasis on risk-based categorization.
MDR: Introduced more detailed classification rules and a stricter risk-based approach, resulting in higher classification for some devices (e.g., software and reusable surgical instruments).
4. Clinical Evaluation Requirements
MDD: Allowed more reliance on equivalence to existing devices.
MDR: Imposes stricter requirements for clinical evaluations and post-market clinical follow-ups, requiring more robust clinical data.
MDD: Fewer and less strictly monitored notified bodies.
MDR: Fewer notified bodies due to more rigorous designation criteria, increasing scrutiny on assessments and certifications.
6. Technical Documentation
MDD: Required technical files but with less stringent details.
MDR: Demands more comprehensive technical documentation, including safety and performance analysis, risk management, and post-market surveillance plans.
7. Post-Market Surveillance
MDD: Basic requirements for post-market surveillance.
MDR: Detailed requirements for proactive post-market surveillance, including Periodic Safety Update Reports (PSURs) for all devices except Class I.
MDR: Mandates UDI for better traceability and monitoring.
9. EUDAMED Database
MDD: Minimal use of centralized databases.
MDR: Introduces the European Database on Medical Devices (EUDAMED) to improve transparency and access to device data.
10. Transitional Period
MDD: Has been phased out.
MDR: Fully applies from May 26, 2021, with transitional provisions for certain devices until May 2024 or 2028, depending on certification status.
The MDR represents a significant overhaul aimed at addressing modern challenges, enhancing patient safety, and ensuring robust oversight of medical devices in the EU.
Importance of Conducting an MDD to MDR Gap Analysis
Conducting a detailed analysis regarding the new MDR requirements is essential. For instance, a change relative to the classification generally implies extra documentation requirements, additional clinical investigation, and a comprehensive risk management review.
It is crucial to consider the timing when conducting a product portfolio assessment, since satisfying any new requirements is not insignificant. Existing design dossiers or technical files must be up to date with the additional mandated requirements when maintaining current MDD certificates past the effective date of the MDR. New application files submitted to the Notified Body after the MDR effective date must meet current MDR requirements along with quality system certification to the new MDR.
Also, additional clinical data may be mandatory depending on the status of the current product portfolios, so include the planning and implementation in your timeline. Toward the beginning of 2021, maintenance of the documentation to the MDR standards will be necessary, while the products that remained certified to MDD will also need to be recertified to MDR.
MDD to MDR Transition: A Key Business Strategy
It is imperative for all medical device manufacturers, as part of the business strategy, to concentrate on accessing their product portfolios and prepare a plan for the timely transition to the MDR. There are limitations worth considering when choosing to commercially distribute legacy medical devices under the old regulatory framework of the MDD. There must be no significant changes – including labeling and packaging changes, which are quite common.
Manufacturers must recognize that not all design changes are foreseeable or predictable, and could likely require recertification to the MDR. For instance, the manufacturer receives a new reportable adverse event requiring a significant labeling update. Maintaining compliance to the MDD EC Certificate after June 2020 would prohibit implementation of this change. Yet, based on this adverse event, the Competent Authority may request corrective action on the labeling change, which would now suddenly propel the company into the MDR transition.
Plan Your EU MDR Transition: Conduct a Gap Analysis with MAVEN
If you are in the midst of planning your EU MDR transition timeline strategy, need help deciphering the requirements, or want any consultancy MAVEN can help. We can conduct a comprehensive EU MDR gap analysis and formulate a clear EU MDR transition strategy and/or train your team.
Why Choose Us?
Expertise: Decades of experience in medical device compliance.
Tailored Solutions: Customized strategies for your unique challenges.
Seamless Support: Dedicated assistance at every stage of the transition.
Start your EU MDR transition with confidence. Contact us today to schedule your gap analysis!