Post-Market Clinical Follow-Up (PMCF) is a continuous process of collecting data from the user/ on humans of CE marked medical devices which is placed on the market or put into service with the aim of confirming its safety and performance throughout its lifetime. As per Annex XIV part B of EU MDR, PMCF process should be carried out by the manufacturer and specified in PMS plan and the findings of PMCF shall be specified in the PMCF report.
The MDCG 2020-7 post-market clinical follow-up (PMCF) Plan template assists manufacturers in complying with the requirements of the MDR for PMCF plan preparation. PMCF information from the device being evaluated should be used to show ongoing performance and safety of the device. The aim of the study is as follows.
What is Post-Market Clinical Follow-Up (PMCF)?
Post-Market Clinical Follow-Up (PMCF) is a process used to gather clinical data on a medical device after it has been placed on the market. It aims to ensure the device’s continued safety, performance, and effectiveness during its lifecycle. PMCF activities include monitoring adverse events, conducting clinical studies, and analyzing real-world data from users. This ongoing evaluation helps identify any potential risks, provides additional clinical evidence for regulatory compliance, and supports product improvements. PMCF is a key part of post-market surveillance and is required by regulatory bodies like the European Medicines Agency (EMA) and CDSCO for medical devices.
Importance of PMCF under EU MDR
Under the EU Medical Device Regulation (MDR), Post-Market Clinical Follow-Up (PMCF) is crucial for ensuring ongoing safety and performance of medical devices after they reach the market. PMCF helps manufacturers collect real-world clinical data to identify and mitigate risks not detected during pre-market clinical trials. It ensures that devices continue to meet the required safety and effectiveness standards throughout their lifecycle. The EU MDR mandates regular PMCF activities as part of a robust post-market surveillance system. This process supports regulatory compliance, helps manufacturers address emerging issues, and provides evidence for the continued marketability of medical devices.
Seven sections that collectively constitute a standard PMCF plan
In compliance with MDCG 2020-7, the plan has to include PMCF study details, such as the PMCF Plan number, date, version, and revision history. The following are the seven sections that collectively constitute a standard PMCF plan.
Section A: Manufacturer contact details
Information about the manufacturer, such as their name and location, phone number, and SRN, should be added to this section. If the manufacturer is located outside the European Union, the MDCG 2020-7 mandates that the name and address of the authorized representative be included in the PMCF Plan.
Section B: Medical Device description and specification
This section should have Product/ trade name, device model or type, general description of device, its intended purpose, intended user, patient population, medical conditions in which it is used, expected lifetime of device, indications, contraindications, and warnings. If the device has any variants, configurations, and accessories, that information must be mentioned in PMCF Plan. If a device is CE certified, then its certification number shall be specified. The applicable CND code, as well as the device class, must be specified in accordance with EU MDR 2017/745 ANNEX VIII Chapter III, Classification Rules. If the product has any novel features, then its novelty in terms of clinical procedures, its features need to be explained in detail.
Section C: Activities related to PMCF – general and specific methods and procedures
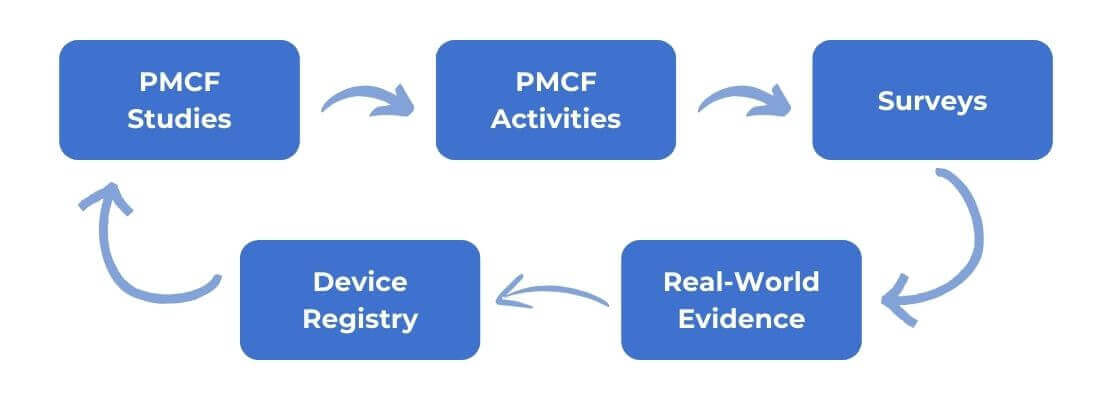
The various post-market activities for the products covered by the PMCF scope are expected to be described in this section, along with general and specific methods, the purpose of each method/activity, and the rationale for the suitability of the activity in achieving specified goals as well as known limitations of the planned activities, for example, incomplete follow up, any missing data need to be mentioned. The timelines of those activities need to be defined.
Device Registry: It is specific for the type of device or the group of the medical devices. Any possible evaluation of suitable national public registries with clinical data on the manufacturer’s own device and/or on similar devices based on the risk of the device(s) and the associated accessories could be specified in this section.
PMCF Studies: It is planned study needs to be specified along with a plan summary including the design, sample size, endpoints, and inclusion/exclusion criteria (e.g. prolonged patient follow-up from pre-market clinical trials, newer clinical studies related to the intended purpose of the device, and retrospective research study). In case of implantable and class III devices where clinical investigations have not been performed pursuant to Article 61(4), the PMCF plan shall include post market studies to confirm the safety and performance of the device.
Surveys: Collect information about the use of the concerned medical device.
Real-world evidence: (RWE) analysis together with a plan summary should have design, sample size, endpoints, and analysis population. The real-world data (RWD) from which these analyses are based should be of sufficient quality and come from reliable data sources.
MDCG 2020-7 requires the justification for the suitability of the selected procedures/ methodologies, including the justification of sample size, timeframes, endpoints, and comparator, all of which should be based on the intended purpose and state of the art. Lastly, study design need to be justified on the basis of all of the above and a statistical justification for acceptability of result in light of the residual risks need to be considered.
Section D: Reference to the relevant parts of the technical documentation
In this section, the manufacturer is required to include references to the relevant information from the clinical evaluation report and from the risk management file, which need to be analyzed, followed up, and evaluated in this plan. Alternatively, the manufacturer must declare that this strategy will not take into account any pertinent data from the risk management file or the clinical evaluation report.
Section E: Evaluation of clinical data relating to equivalent or similar devices
The manufacturer shall specify information regarding equivalent/similar devices for which clinical data will be further evaluated and presented with clear reference in the PMCF report. Data from equivalent or similar devices may be used, only for updating the information relating to the state of the art, to identify and further assess relevant safety outcomes etc. The product name of each equivalent or similar device, along with its intended use, intended users, intended patient population, medical condition, indication, and reference to the clinical data evaluation in the CER (including its date, version, and location in the text), should be provided in this section.
Section F: Reference to any applicable common specification(s), harmonized standard(s) or applicable guidance document(s)
Any common specifications, harmonized standards, and PMCF guidance which apply to PMCF study need to be specified.
Section G: Estimated date of the PMCF evaluation report
When the manufacturer plans to have the first report, shall be stated at the end. The timelines for the same shall be defined quarterly or at least yearly.
Challenges in PMCF Documentation and Maintenance
Challenges in PMCF documentation and maintenance include ensuring comprehensive and accurate data collection over the device’s lifecycle. Manufacturers must track real-world performance and adverse events, which can be resource-intensive and time-consuming. Maintaining up-to-date records for regulatory compliance requires careful coordination across teams, especially when dealing with complex devices. Data analysis and reporting must be precise to identify emerging risks or trends. Additionally, coordinating clinical follow-up activities across multiple regions, dealing with varying regulatory requirements, and addressing incomplete or inconsistent data pose significant hurdles. Effective documentation and maintenance require robust systems, continuous monitoring, and timely updates to ensure compliance and safety.
References
1. EU MDR 2017/745: REGULATION (EU) 2017/745 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 5 April 2017
2. MDCG 2020-7: Post-market clinical follow-up (PMCF) Plan Template A guide for manufacturers and notified bodies