What is IVDR and Why is it Important?
There has been an EU Directive (98/79/EC) regulating in vitro diagnostic medical devices (IVDs) since 1993. However, problems emerged with the interpretation and application of that Directive, particularly that it provided only low levels of scrutiny for potentially ‘high risk’ devices.
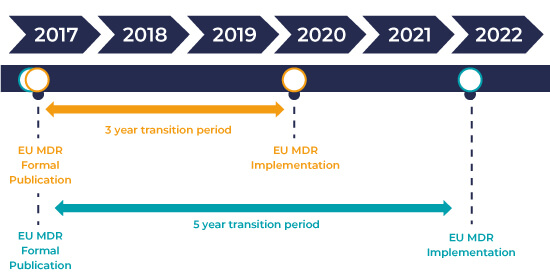
IVDR Transition Timeline and Implementation
The IVDR came into force on 26 May 2017 and has a 5-year period of transition. This transition period for IVDR regulation ensures sufficient time for manufacturers and developers to utilize new pathways to market and for suppliers to adapt existing IVDs to comply with new requirements of IVDR.
- Quality, safety and reliability of IVDs will be improved by the IVDR.
- The IVDR gives new definitions of what an IVD is and seeks to improve how IVDs are classified and how IVDs are kept under surveillance and regulated in IVDR.
- IVDR also consider and regulate certain, algorithms as IVDs which satisfthe definitions mentioned in IVDR regulation.
- There are some challenges associated with how algorithms will be regulated by the IVDR.
“Each IVD currently on the market must be transitioned in new system Check if instrument or product is still or becomes (part of) an IVD”
The European Commission’s new In Vitro Diagnostic Regulation (IVDR 2017/746) will address several weaknesses of the IVDD and bring significant regulatory changes for IVD manufacturers selling in Europe. The IVDR will apply from 26 May 2022, but proactive IVD companies are planning their implementation strategies for IVDR.
Key Changes in IVDR & Classification Rules
- The definition of IVDs in the IVDR includes devices that are ‘software or system’ where used in combination with the examination of specimens including blood and tissue for the purpose of obtaining in-vitro information concerning ‘predisposition to a medical condition or a disease’ or used to ‘predict treatment response or reactions’ (Article 2(2)), any of which could use algorithms.
- The IVDR introduces new classification rules based on the Global Harmonization Task Force System with four-risk based classes – Class A (low) to Classes B, C and D (highest risk). Under the current Directive most standalone algorithms that have a medical purpose and qualify as an IVD are simply required to be CE marked via self-assessment by the developer or manufacturer (i.e. the lowest-classification with light-regulation in Class A). Under the IVDR Classifcation, most IVDs including algorithms, will ‘up-classify’ into the higher risk Classes B, C and D requiring the involvement of Notified Bodies (independent certification bodies) rather than simple self-assessment.
IVDR Classification and Its Impact on the Industry
Concerns have arisen in some medical IVD areas, including genetic and genomic testing, as to the ability of the new IVDR to better regulate use of such algorithms. The IVDR may have an impact on the genetics/ genomics sector in several ways:
- All human genetic tests are brought within the scope of the IVDR and classified as Class C IVDs based on the IVDR Classification.
- Clinical genomic services are increasingly reliant on algorithms to power technologies such as whole genome sequencing, and interventions such as risk stratification that rely on algorithms combining multiple genetic and non-genetic factors to make personalized risk predictions .
- The IVDR will impose more stringent requirements on test developers and manufacturers for conformity assessment and surveillance
- Significant questions remain about how validation and replication will be proportionately regulated in IVDR.
Impact of IVDR on IVD Companies
- 2000 references in the scope (IVD CE MARKED).
- 15 sites (worldwide).
- 15 departments involved.
- 5 Years transition.
- From 3 to 73% of IVDs with involvement of Notified Bodies in the CE Marking process
IVDR PERFROMANCE EVALUATION
- Performance Evaluation is mandatory (Chapter VI of IVDR).
- Post market performance follow-up plan is mandatory as per Annexure XII of IVDR).
- For class C & D yearly updates required (Annexure XII of IVDR).
- Scientific and ethical approval necessary for performance studies (Chapter VI of IVDR).
EU will require that all products on the market are phased into the new system by the end of Transitional period.
This means that you have to do a new conformity assessment under the new IVDR regulation for all the devices currently on market or remove the product from the market.
Achieve IVDR Compliance with Maven Profcon’s Expert Solutions
Navigating IVDR requirements can be complex, but Maven Profcon simplifies the process. Our expert team ensures seamless compliance through tailored strategies, efficient documentation, and regulatory support. With our proven track record, we help IVD manufacturers transition smoothly while meeting stringent EU standards. Trust us for reliable IVDR compliance solutions.