What is In Vitro Diagnostic (IVD) Medical Device?
In Vitro Diagnostic (IVD) Medical Device means any medical device intended by the manufacturer to be used in vitro for the examination of specimens, including blood and tissue donations, derived from the human body, to provide information regarding a physiological or pathological process or state, concern congenital physical or mental impairments, concerning the predisposition to a medical condition or a disease, to determine the safety and compatibility with potential recipients, to predict treatment response or reactions, or to define or monitor therapeutic measures.
IVD medical devices can be a reagent, reagent product, calibrator, control material, kit, instrument, apparatus, piece of equipment, software, or system, whether used alone or in combination. For example, the most common IVD medical devices that we all are aware of are the testing kit for Covid-19, Pregnancy Test Kit, and a glucometer.
Why IVDR (EU) 2017/746 Matters for Device Safety
There was a “fallout” due to the breast implant scandal, where a manufacturer deliberately used industrial-grade silicone to make breast implants. Hence, moving from a Directive to a Regulation was essential. The aim was to ensure broad scope of protection, more effective implementation of the rules, and to provide transparency.
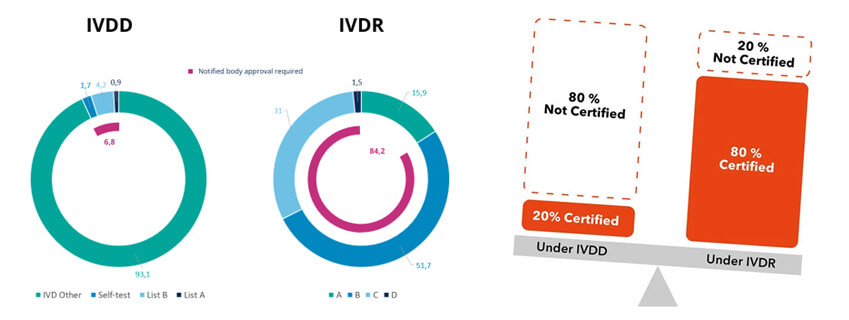
The scope of the IVDR regulation has not expanded notably. The range of products covered by the IVDR has remained the same; however, the depth and the expectations for the documentation and review have significantly increased.
REGULATION (EU) 2017/746 (IVDR) aims to ensure a high level of protection of health of patients and users by setting high standards of quality and safety for in vitro diagnostic medical devices.
The most significant change in IVDR is the classification of devices. Where the devices are divided into two lists i.e., List A and List B in IVDD, the devices in the IVDR are divided into four classes i.e., Class A, Class B, Class C, and Class D. Class D devices have the highest risk while Class A devices have the lowest. The devices are further categorized as devices for near-patient testing, devices for self-testing, and companion diagnostic devices.
Suggested Read:
Cdsco New Risk Classification For Medical Devices
What is a “significant change” under the EU MDR or IVDR?
A “significant change” under the EU MDR or IVDR refers to any modification in an IVD device that could impact its safety, performance, intended use, or regulatory compliance. Changes requiring reassessment under the IVD regulation include alterations in the design, materials, manufacturing process, software, or intended purpose of the IVD devices. Additionally, modifications that could affect the risk classification or require updated clinical or performance evaluations are also considered significant.
For manufacturers holding an IVD certificate, any significant change may necessitate a new conformity assessment or re-certification under the IVD regulation. This ensures that the updated IVD devices continue to meet safety and performance requirements. Notified Bodies play a crucial role in determining whether a change qualifies as significant. It is essential for manufacturers to assess and document any changes carefully to maintain compliance with IVDR and avoid disruptions in market access.
By proactively managing design and process modifications, manufacturers can ensure that their IVD devices remain in compliance while minimizing regulatory hurdles.
IVDR Transition Periods for Legacy and Old Devices
IVDR has been effective since May 26, 2017. As it is applicable within the territory of the European Union, it is necessary for any medical device manufacturer that is going to distribute its products in the European market to comply with the appropriate regulations.
However, for medical devices that are already present in the market, special transition periods are provided. These devices can be defined by two terms: “Legacy Devices” and “Old Devices”. So, first let’s get a clear understanding of the terms.
Legacy Devices under IVDR
Legacy Devices are the devices covered by a valid EC certificate issued by a notified body in accordance with Directive 98/79/EC on in vitro diagnostic medical devices (IVDD) prior to 26 May 2022; or the devices for which a declaration of conformity was drawn up prior to 26 May 2022 in accordance with the IVDD and for which the conformity assessment procedure pursuant to the IVDR (contrary to the IVDD) requires the involvement of a notified body.
Old Devices Under IVDR
The devices that were placed on the market or put into service before 26 May 2022 in accordance with the IVDD or the applicable national rules before the IVDD had become applicable and which are still on the market or in use after 26 May 2022.
So, now that you have understood the terms. Let’s move forward with the Transition periods: –
For devices covered by a valid EC certificate issued in accordance with the IVDD (i.e., devices listed in Annex II IVDD and devices for self-testing), prior to 26 May 2022, the transition period ends on 26 May 2025.
If placed on the market before 26 May 2025, those devices may continue to be made available until 26 May 2026.
As per the class of the device:
Devices for which a declaration of conformity was drawn up prior to 26 May 2022 in accordance with the IVDD and for which the conformity assessment procedure pursuant to the IVDR (contrary to the IVDD) requires the involvement of a notified body, the transition period ends on:
Even though the device may continue to be made available in the market as per the timelines, still there are some of the essential requirements of IVDR for legacy devices and old devices that needs to be met.
When does a CE-marked device (medical device or IVD) qualify as legacy device?
A CE-marked medical device or IVD qualifies as a legacy device if it was placed on the market under the IVDD (In Vitro Diagnostic Directive) 98/79/EC or the MDD (Medical Device Directive) 93/42/EEC, before the transition to IVDR (EU) 2017/746 and MDR (EU) 2017/745. Legacy devices include those with valid EC certificates issued before May 26, 2022, and those that now require a Notified Body’s involvement under IVDR. However, these devices must comply with essential post-market surveillance, vigilance, and registration requirements. Manufacturers of IVD diagnostics must ensure a smooth transition from an IVDD device to IVDR compliance to continue selling their products in the EU market.
Transition Periods for Legacy and Old Devices
1.Transition Timeline:
- Legacy devices with valid MDD/AIMDD certificates can remain on the market until the certificate’s expiration or May 26, 2024, whichever is sooner.
- Devices must meet MDR requirements related to post-market surveillance, vigilance, and registration.
2.Extended Transition for Certain Devices: Amendments allow an extended timeline for higher-risk devices until May 2027 and lower-risk devices until May 2028, under specific conditions.
3.Old Devices: Devices without valid MDD/AIMDD certificates cannot be marketed after the transition unless they are re-certified under MDR.
4.Prerequisites for Transition
- Compliance with MDR’s post-market requirements.
- No significant changes to the device design or intended use.
5.EUDAMED Registration: Legacy and old devices must be registered in EUDAMED for continued compliance.
6.Risk Mitigation: Manufacturers must maintain robust quality management systems and risk management practices to ensure uninterrupted market availability.
Application of IVDR requirements to ‘Legacy Devices’:
In addition to the requirements set out above, also other IVDR requirements should apply to ‘legacy devices’, provided that those requirements relate to post-market surveillance, market surveillance, vigilance or registration of economic operators and devices and support a well-functioning vigilance and market surveillance system as well as proper registration of economic operators and devices.
Regarding ‘legacy devices’ covered by certificates issued under the IVDD, the notified bodies that issued the respective certificates conduct the ‘appropriate surveillance’, which essentially is a continuation of the previous surveillance activities under the IVDD. In the framework of their surveillance activities, notified bodies will take into account the new requirements that apply to manufacturers resulting from the transitional provisions.
Application of IVDR requirements to ‘Old Devices’:
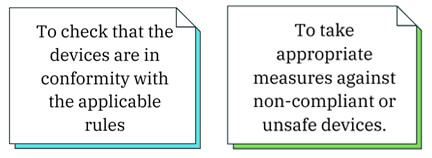
- IVDR requirements are in principle not applicable to ‘old’ devices. However, Articles 88 to 95 IVDR, which lay down rights and obligations of competent authorities with regard to market surveillance activities, apply to ‘Old Devices’ after 26 May 2022.
- Moreover, the reporting and analysis of serious incidents and field safety corrective actions occurring after 26 May 2022 should be done in accordance with Article 82 and 84 of IVDR.
- This allows competent authorities:
While this new regulatory environment can improve patient satisfaction and the accuracy of treatment decisions, in the long run, the development of in vitro diagnostic devices will not be as smooth as it once was. From stricter documentation processes to the involvement of Notified Bodies and the associated additional costs, stringent compliance requirements and guidelines will place a heavy burden on IVD manufacturers.
Manufacturers of in vitro diagnostics who wish to sell their products in the European market, need to fulfill the requirements of IVDR to be fully compliant, and now will be the time to get ready. If the preparation is put off until the end of the transition phase, there are chances of experiencing many challenges. So, start now and if you need any assistance, contact us, we would be happy to cater to your needs.
How Maven can help you with this?
- We are a One-Stop Solution for IVD CE Marking – Our In-vitro Diagnostic Device CE Marking Service covers: General IVDs, Self-testing IVDs and all other classes of devices ensuring that quality certification is provided to the manufacturer to legally sell their products in the European Union.
- Whether it’s Gap Analysis and compliance of legacy/old devices with IVDR or preparation and compilation of technical files to application and procuring CE mark of conformity for your In-Vitro Diagnostic (IVD) device by complying with the applicable In-Vitro Diagnostic Regulations we will assist you at every step.
- We take extra care to maintain control over documentation and client confidentiality which creates value and helps us maintain client’s best interests.
- Error free submission and Expert advice.
Author: Divyanshi Jethva