EUDAMED is the European Database on Medical Devices, developed under the EU Medical Device Regulation (MDR 2017/745) and In Vitro Diagnostic Regulation (IVDR 2017/746) to improve transparency, coordination, and safety across the EU. It serves as a central platform for collecting and sharing information on medical devices, including registration, clinical investigations, vigilance, and market surveillance. Unlike its predecessor, Eudamed2, which was limited to regulatory authorities, the new EUDAMED is publicly accessible and multipurpose. It functions as a registration, collaboration, notification, and dissemination system, and supports data interoperability among stakeholders. It covers all finished medical devices (excluding custom-made ones) and aims to enhance oversight while improving patient and user confidence in the EU market. By centralizing data, EUDAMED plays a vital role in ensuring safer, more transparent regulation of medical devices across Europe.
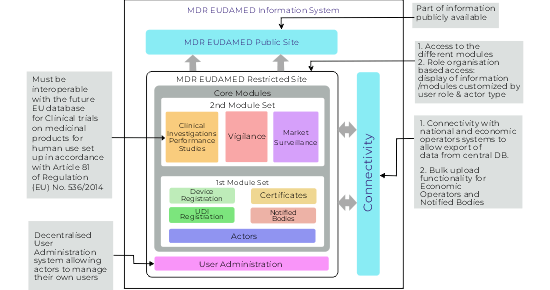
EUDAMED is designed as a comprehensive and integrated database that supports the implementation of the EU Medical Device Regulation (MDR) and In Vitro Diagnostic Regulation (IVDR). It consists of six interconnected modules, each serving a specific regulatory function.
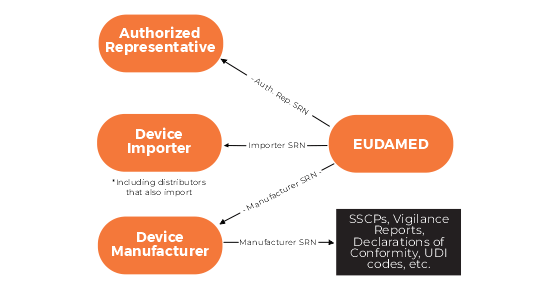
- Actor Registration Module – This allows economic operators (manufacturers, importers, authorised representatives) to register and obtain a Single Registration Number (SRN), a key identifier for regulatory processes.
- UDI/Device Registration Module – Manufacturers must register their devices using the Unique Device Identification (UDI) system, which improves traceability and facilitates recalls and safety alerts.
- Notified Bodies and Certificates Module – Stores information about notified bodies, issued certificates, and their scope, improving transparency of conformity assessments.
- Clinical Investigations and Performance Studies Module – Centralises data on ongoing and completed clinical investigations for medical and in vitro diagnostic devices.
- Vigilance and Post-Market Surveillance Module – Enables reporting and analysis of serious incidents, field safety corrective actions, and other vigilance activities.
- Market Surveillance Module – Supports coordination between national competent authorities for regulatory enforcement and oversight.
Together, these modules make EUDAMED a powerful tool for improving transparency, coordination, and safety in the regulation of medical devices throughout the EU.
Implementation Timeline and Key Deadlines
The implementation of EUDAMED under the EU Medical Device Regulation (MDR 2017/745) and In Vitro Diagnostic Regulation (IVDR 2017/746) is being rolled out in phases to ensure smooth adoption. Although the original full functionality was planned for 2020, delays have led to a new phased timeline.
The Actor Registration Module is already operational and available for use. Additional modules—such as UDI/device registration, notified bodies and certificates, clinical investigations, vigilance, and market surveillance—will be released once all six modules are fully functional and audited.
The European Commission has stated that EUDAMED will become mandatory 6 months after a formal notice of full functionality is published, expected in 2026. Until then, the use of available modules remains voluntary but highly encouraged.
Manufacturers should not wait for full deployment—early registration and preparation are essential to ensure compliance and smooth integration once the database becomes fully operational.
Benefits of EUDAMED for Stakeholders
EUDAMED offers significant benefits to all stakeholders involved in the regulation and use of medical devices across the EU. For manufacturers and economic operators, it streamlines compliance by centralizing device registration, vigilance reporting, and certificate management, reducing administrative burdens and improving traceability. Regulatory authorities gain a powerful tool for coordinated market surveillance, enabling quicker responses to safety issues and improved oversight.
For notified bodies, EUDAMED simplifies communication and documentation sharing with manufacturers and authorities. Healthcare professionals and the public benefit from increased transparency, as the database provides open access to key information about devices, such as safety notices, clinical data, and certifications.
Overall, EUDAMED enhances patient safety, fosters regulatory harmonization, and supports informed decision-making across the EU medical device ecosystem. Its interoperable design ensures seamless data exchange and collaboration, strengthening trust and accountability among all stakeholders involved.
Data Privacy and Confidentiality in EUDAMED
EUDAMED is designed with strong data privacy and confidentiality safeguards in line with the EU’s General Data Protection Regulation (GDPR). While the system promotes transparency, it also ensures that sensitive information—particularly personal and proprietary business data—is adequately protected. Access levels are carefully controlled based on the role of the user, such as manufacturers, notified bodies, competent authorities, or the public.
Publicly available data, such as device registration details and safety notices, helps improve patient awareness and trust. However, confidential information, including internal clinical data or trade secrets, is restricted to authorized users only. Data transmission within EUDAMED is secured through encryption, and user access is logged and monitored to prevent misuse.
By balancing openness with stringent privacy controls, EUDAMED ensures compliance with data protection laws while supporting effective device regulation, post-market surveillance, and patient safety across the EU. Manufacturers must also ensure their data submissions meet these standards.
The EU MDR 2017/745 establishes an EU database to enhance overall transparency related to economic operator, registration, clinical investigation, vigilance, market surveillance reporting by the deadline of November 26, 2022. The EUDAMED database will cover all finished good medical devices and only excludes custom-made devices from data entry.
The new regulations on medical devices (EU MDR) and on in vitro diagnostic medical devices (IVDR) establish a much wider EUDAMED database than the existing one under the current directives (Eudamed2). Currently, the EC database on medical devices, Eudamed2, is a secure web-based portal. It is a central repository for information on market surveillance exchanged between national competent authorities and the Commission. Its use is restricted to national competent authorities, it is not open for consultation and is not publicly accessible.
However, the new medical devices regulations (EU MDR) contain important improvements including a much larger EUDAMED database. The new EUDAMED will be multipurpose. It will function as a registration system, a collaborative system, a notification system, a dissemination system (open to the public), and will be interoperable.
Commission’s Present Situation on The Implementation of The EUDAMED Database
The European Commission is delaying the launch of Eudamed for both medical devices and in-vitro diagnostics to May 2022, it was confirmed recently that Eudamed cannot be made operational until the entire system and its different modules have achieved full functionality and are subject to an independent audit. The date of application of MDR still remains May 2020, which effectively means that device manufacturers can postpone their entering of data into the Eudamed database.
Structure of New Eudamed:
As per Functional specifications for the European Database on Medical Devices, first draft version the 4.1, MDR Eudamed Information system would consist of two components.
- MDR EUDAMED public site
- MDR EUDAMED restricted site
The public site of Eudamed will have part of the information to be publicly accessible, it is not clear, what exact information will be available and accessible to the public. Restricted site of Eudamed, grants access to different modules and it has role-organization based access, which means a display of the information or modules will be customized by user roles and actor types. As connectivity is concerned with national and economic operators’ systems allows the export of the data from the central database to a local database. It also offers bulk upload functionality for economic operators and notified bodies. Decentralized user administration will allow actors to manage their own users.
Suggested Read: Responsibilities of Economic Operators as per EU MDR 745/2017
The below table outlines the additional information requiring submission into Eudamed database as per annex VI.
| Subject | Details |
| Economic Operator | Contact information and SRN for manufacturer, authorized representative, or importer/distributor |
| Regulatory Contact | Company regulatory representative or third-party submitter in charge of submission |
| Unique Device Identifier (UDI) | Basic UDI-DI; May also need to include UDI-PI (the production identifier) in the form of an expiration date, manufacturing date, lot number, or serial number; Basic UDI-DI shall appear on the Declaration of Conformity |
| Regulatory approval details | Includes the approval certificate number and Notified Body details |
| Country availability | Member State where device will be placed on the market in the EU (availability for class IIa, IIb, and III devices) |
| Device risk classification | I, IIa, IIb, III |
| Single use Notice | Denote if device is a reprocessed single use device (labeled as single use device or list maximum number of reuses). |
| Drugs/Possible Contaminants | Presence of medicinal drug/product, human blood/plasma derivative, tissues/cells of human/animal origins, latex, or phthalates |
| Clinical Investigation | Identification number of clinical investigation(s), if applicable |
| Intended purpose | Intended purpose if other than medical |
| Public statement on safety | Summary of safety and clinical performances (class III or implantable devices only) |
| Device status | On the market, no longer placed on the market, recalled, field safety corrective action details, or other |
| Quantity | Quantity per package |
| Medical Device Nomenclature Code | Internationally recognized medical devices nomenclature is available free of charge and made available by the EU regulatory commission – Italy’s National Classification of Medical Devices (CND) mapped to Global Medical Device Nomenclature (GMDN) codes |
| Product Identification | Product trade name, description, and number (catalogue/reference code) |
| Clinical Product Size | Volume, length, diameter, or other measurement |
| Storage and Handling Condition | Details on how the device should be stored and handled |
| Sterility | Details on sterility or need for sterilization prior to use |
| Electronic IFU | URL for additional information such as eIFU (optional) |
| Vital Safety Information | Critical warnings, precautions, or contra-indications, if applicable |
The new Eudamed database is much larger than the existing Eudamed2 database. With extended timelines of Eudamed implementation, it will be interesting for manufacturers to go through the transition period after EU MDR comes into the effect in May 2021 and to be prepared for the Eudamed launch.
EUDAMED Myths And Misinformation : –
Myth: The EC will reverse its decision not to use GMDN codes.
- CND is going to be the Eudamed nomenclature, this is decided and this fact will not change. This is a fact and the relevant decision has been officially published. The mapping exercise between GMDN and CND either has started or will start shortly but this CND decision is final.
Myth: The EC is going to provide us with all the support we need, our IT teams can just contact them.
- The EC will provide manuals, FAQ’s, various IT targeted publications (e.g. Data Dictionary, XSD, etc.), and a functional mailbox where people can submit questions. They will not provide a call center. Manufacturers with technical issues relating to failed uploads, XML not validating against the XSD, or data dictionary questions, will not have EC support people at the end of a phone waiting to assist them. This will simply not happen; the costs of such call center would be too big a burden. Companies will have to rely on their own IT experts and/or external support companies to answer such questions. Providing IT support is not the role of the EC.
Myth: We can apply to our Competent Authority in advance for a SRN.
- This is completely wrong. The SRN is an Eudamed generated alphanumeric string that will be available to each registering company after they have registered in Eudamed and have been successfully validated by their competent authority (CA). This CA validation and SRN creation can only happen on or after March 26th 2020, when Eudamed goes live.
Myth: The SRN will be the same format as our VAT number.
- This is another incorrect piece of information. In the XSDs published by the EC in June 2019, the format of the SRN is included. This is the format of the SRN:
Length 15 characters
Format: ISO Country Code-Actor Type-9 digits
Actor type example: Manufacturer: MF, Importer: IM
Full code: IE-MF-000000001
Suggested Read: MDR 2017/745: Responsibilities and Authorities Decoded
EUDAMED Delay :-
One of the greatest, time sensitive, bottlenecks was always going to be actor registration. Under the initial March 2020 go-live plan, competent authorities (CA) were always going to be under severe pressure to complete validations.
The main benefit is the manufacturers, system / procedure pack producers, authorized representatives, and importers, will be able to register and receive their SRN well in advance of the May 2022 deadline. They will be under no pressure to get the SRN, required to be able to get their devices and vigilance issues registered in Eudamed. The EO’s will be able to register, and wait with no time pressures, comfortable in the knowledge the CA’s are working on their registration validation.
This is even better news for the Non-EU manufacturers who were even more disadvantaged with registration process. Non-EU manufacturers could not register until their authorized representative had completed their registration. A non-EU manufacturer has a double validation, firstly by their authorized representative and secondly by the CA.
The EUDAMED is just one examples of many new demands being placed by the new European Medical Device Regulation. It can be difficult to make sense of it all but there are ways we can help.
We’d be glad to help!!
Kindly Contact:
MAVEN PROFCON SERVICES LLP