Decoding the First-Ever MDCG Guidelines for Annex XVI Products!
The newly proposed MDCG guidelines are a valuable resource for manufacturers (of products without an intended medical purpose), notified bodies, and stakeholders navigating the intricate landscape of medical device regulatory standards. A comprehensive review of these guidelines is crucial to maintain the safety and efficacy of medical devices in the market.
On 14th December 2023, the Medical Device Coordination Group (MDCG) has released two new documents: MDCG 2023-5 (Guidance on qualification and classification of Annex XVI products) & MDCG 2023-6 (Guidance on demonstration of equivalence for Annex XVI products)
What is Annex XVI of EU MDR?
Annex XVI of EU MDR 2017/745 outlines six categories of products which are governed by EU MDR even though they lack an intended medical purpose. The European Commission determined that products falling within these categories, for which manufacturers claim only aesthetic or non-medical purposes but exhibit similarities to medical devices in terms of functionality and risk profile, should be covered by the EU MDR. Following are the examples for Annex XVI Products :
Examples for Annex XVI Products
| SR NO. | GROUP | EXAMPLES |
| 1 | Contact lenses or other items intended to be introduced into or onto the eye. | Non-Prescription Colour Contact Lenses |
| 2 | Products intended to be totally or partially introduced into the human body through surgically invasive means for the purpose of modifying the anatomy or fixation of body parts with the exception of tattooing products and piercings. | Solid body contour modifying implant (e.g.
breast implants) |
| 3 | Substances, combinations of substances, or items intended to be used for facial or other dermal or mucous membrane filling by subcutaneous, submucous or intradermal injection or other introduction, excluding those for tattooing | Dermal fillers |
| 4 | Equipment intended to be used to reduce, remove or destroy adipose tissue, such as equipment for liposuction, lipolysis or lipoplasty. | Body shaping equipment |
| 5 | High intensity electromagnetic radiation (e.g. infra-red, visible light and ultraviolet) emitting equipment intended for use on the human body, including coherent and non-coherent sources, monochromatic and broad spectrum, such as lasers and intense pulsed light equipment, for skin resurfacing, tattoo or hair removal or other skin treatment. | Intense pulsed light (IPL) equipment (e.g. for
hair removal) |
| 6 | Equipment intended for brain stimulation that apply electrical currents or magnetic or electromagnetic fields that penetrate the cranium to modify neuronal activity in the brain. | Transcranial stimulation equipment (surgically non-invasive) for non-medical purposes |
Requirements of Annex XVI Products:
Both the new guidelines are a must-read for the manufacturers of device that fall under Annex XVI medical devices (products without an intended medical purpose).
- New MDCG 2023-5 guideline: Guidance on qualification and classification of Annex XVI products – A guide for manufacturers and notified bodies.
- This guidance document provides elements useful for the qualification of a product as a product without an intended medical purpose listed in Annex XVI to the MDR. It also provides explanations and examples for the application of certain classification rules to products without an intended medical purpose.
- This guidance document should be used in conjunction with the MDCG 2021-24 on classification of medical devices and take into consideration Commission Implementing Regulation (EU) 2022/2347 on reclassification.
In addition, the guidance is particularly focused on dual-purpose devices, addressing the complexities of classifying products that serve both medical and non-medical purposes. It also offers practical advice on interpreting relevant terms and concepts, aiding manufacturers and notified bodies in ensuring regulatory compliance within the EU.
Who Needs to Comply with MDCG Guidelines?
Manufacturers, notified bodies, and stakeholders involved in Annex XVI products must comply with the MDCG guidelines to ensure regulatory adherence under the EU MDR. The MDCG 2023-5 guideline helps manufacturers determine whether their product qualifies as an Annex XVI product and how it should be classified. It also provides clarity on dual-purpose devices, which serve both medical and non-medical purposes, and addresses the regulatory requirements for accessories associated with these products.
On the other hand, the MDCG 2023-6 guideline focuses on demonstrating equivalence for Annex XVI products. It guides manufacturers on using existing data from similar products to meet the CE-marking requirements. For dual-purpose devices, this guidance applies only to their non-medical intended use.
Notified bodies must use these guidelines when evaluating Annex XVI products to ensure compliance with regulatory standards. Additionally, manufacturers should reference MDCG 2021-24 and Commission Implementing Regulation (EU) 2022/2347 for a comprehensive understanding of classification and reclassification rules.
By following these MDCG guidelines, manufacturers can ensure compliance, improve product safety, and facilitate smoother market entry within the EU. These documents play a crucial role in bridging the regulatory gap for products that lack an intended medical purpose but share functional similarities with medical devices.
Additional Crucial points of MDCG 2023-5:
- Accessories for non-medical products aren’t specifically mentioned in Article 2 of the MDR. However, the guidance clarifies that they fall under regulation if they match the description in Annex XVI and fit the definition of a regulated product in the CS. If an accessory can be used with a product, it can be sold as part of that product. If it can be used independently, it can be considered a stand-alone product.
- Dual-purpose devices, claiming both medical and non-medical purposes, must meet the stringent requirements of both the CS (Common Specifications) and the MDR, particularly regarding risk. If simultaneous dual-purpose use is not achieved, apply MDR and CS requirements separately, but consider combining certain measures as they may impact both intended purposes.
- In the case of Multi-Purpose Products, the product may share characteristics with multiple groups of Annex XVI. Ideally, each product should belong to only one of the six groups. But, if the product falls under more than two or more groups, the cumulative requirements of all applicable requirements in the CS shall apply.
Example: Breast Implants
These would be classified as Class III, Rule 8 according to MDCG 2023-5. These fall under section 2 of Annex XVI of EU MDR 2017/745, which states products intended to be totally or partially introduced in the human body through surgically invasive means for the purpose of modifying the anatomy or fixation of body parts.
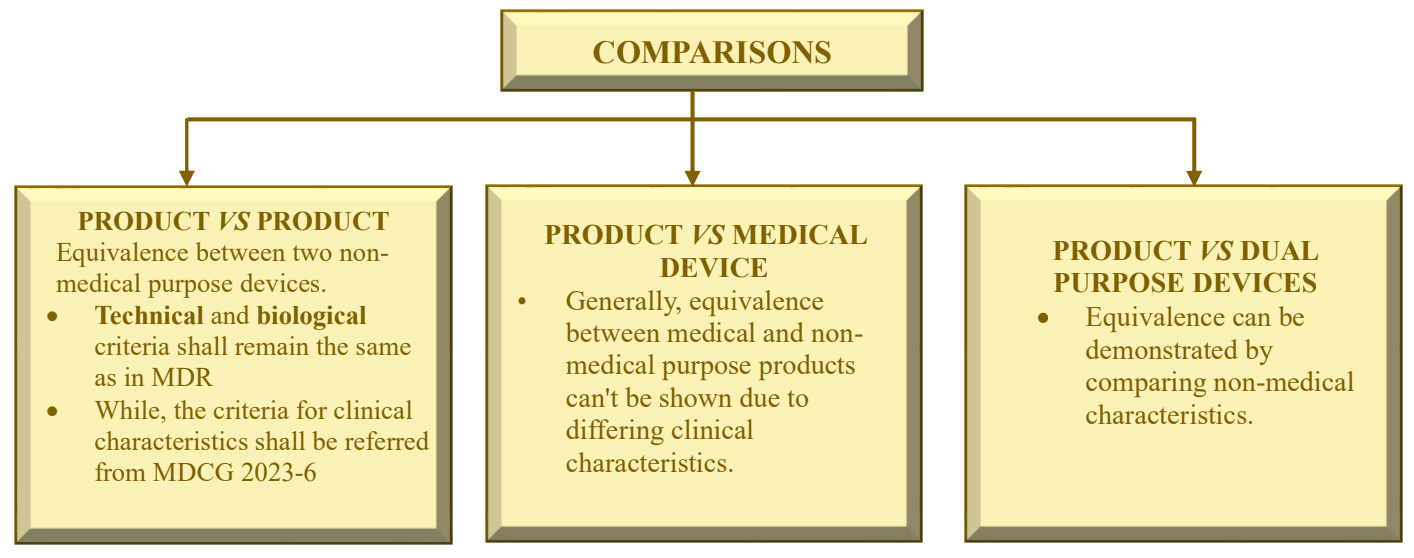
New MDCG 2023-6 guideline
- New MDCG 2023-6 guideline: Guidance on demonstration of equivalence for Annex XVI products – A guide for manufacturers and notified bodies.
- This MDCG guidance covers the demonstration of equivalence, based on data pertaining to an already existing device, for the purpose of CE-marking under the MDR and is applicable to products without an intended medical purpose listed in the Annex XVI of MDR and covered by the CS (Common Specifications). For dual-purpose devices, which are devices with a medical and a non-medical intended purpose, this guidance applies only to the non-medical intended purpose.
- This guidance document should be used in conjunction with MDCG 2020-5 on equivalence.
Impact of MDCG Guidelines on Manufacturers and Market Entry
The new MDCG guidelines, MDCG 2023-5 and MDCG 2023-6, have a significant impact on manufacturers producing Annex XVI products—devices without an intended medical purpose but regulated under EU MDR due to their functional similarities with medical devices. These guidelines introduce stricter classification criteria, requiring manufacturers to thoroughly assess their products based on risk profile, intended use, and compliance with Common Specifications (CS).
For market entry, manufacturers must now demonstrate compliance with these enhanced regulatory requirements, including qualification and classification under MDCG 2023-5 and equivalence demonstration under MDCG 2023-6. This adds an additional layer of scrutiny in obtaining CE marking, making regulatory approvals more complex and time-consuming.
Additionally, the focus on dual-purpose devices presents challenges for companies that manufacture products with both medical and non-medical purposes, as they must meet the requirements of both categories. Manufacturers must also carefully evaluate accessories, as the guidance clarifies their regulatory status under Annex XVI.
Ultimately, these guidelines ensure higher safety and quality standards but demand increased diligence from manufacturers. Companies must invest in regulatory expertise and strategic planning to navigate these requirements efficiently and ensure a smoother market entry process.
Common Pitfalls to Avoid in Annex XVI Product Qualification
When qualifying Annex XVI products, manufacturers often face pitfalls that can lead to regulatory setbacks. A common mistake is misinterpreting the classification criteria, leading to incorrect product categorization. Failing to provide sufficient clinical data to demonstrate safety and performance can also result in compliance challenges. Overlooking dual-purpose device requirements, where products serve both medical and non-medical purposes, may lead to incomplete regulatory submissions. Additionally, neglecting to align with Common Specifications (CS) and MDR standards can delay market entry. To avoid these pitfalls, manufacturers must thoroughly assess classification rules, provide robust data, and stay updated on evolving MDCG guidelines.
Conclusion
In conclusion, when embracing these guidelines, it is crucial to understand the guiding principles and consider the similarities with medical devices.
References:
[1] EU MDR 2017/745
[2] EU 2022/2346
[3] EU 2022/2347
[4] MDCG 2023-5: Guidance on qualification and classification of Annex XVI products
[5] MDCG 2021-24: Guidance on classification of medical devices
[6] Figure-1
[7] MDCG 2023-6: Guidance on demonstration of equivalence for Annex XVI products
[8] MDCG 2020-5: Clinical Evaluation – Equivalence