What Is a Clinical Evaluation?
Clinical Evaluation is the collection, assessment and analysis of clinical data related to medical device to verify its clinical Safety, efficacy and performance. The Clinical Evaluation Report (CER) contains the results of the clinical evaluation and the clinical evidence on which it is based, in order to support the assessment of the device’s conformity.
Clinical evaluation is a continuous or on-going process conducted throughout the life cycle of a medical device based on a comprehensive analysis of available pre-and post-market clinical data related to the intended use of the device in question, including clinical performance and safety data from PMS, Vigilance, PMCF, or clinical investigation.
To achieve regulatory compliance and authorization for sale in Europe, every medical device need to proof its safety, efficacy and performance must be supported by a Clinical Evaluation Report (CER).
This is not a new requirement for medical device manufacturer, but after the introduction of new medical device regulation (MDR) introduced in May 2017 tightened requirements for CERs now there are more Post Marketing Clinical Follow-Ups (PMCF) studies and clinical data require for CERs, should be meticulously scrutinized.
Manufacturer must be able to comply all the requirement as per the MEDDEV 2.7/1 revision 4 June 2016 article 61 and Annex XIV of EU MDR to prepare the Clinical Evaluation Report for Medical Devices. Otherwise they having risk of removal of products from the market, due to reason of medical device not proof its safety, efficacy and performance claimed by manufactures.
Guidelines for Writing a Clinical Evaluation Report
Manufacturer should start writing of Clinical Evaluation Report by collecting, assimilating, and objectively present data about the medical device in accordance with the requirements of MEDDEV 2.7/1. This will require input from other experts, e.g. the manufacturer for technical information about the device, librarian or information scientist for literature searches, quality specialist for complaints data, and safety scientist for PMS & PMCF data.
The objective is to support conformity of the device with the essential requirements for safety and performance as per the European Medical Devices Directive (MDD) 2007/47/EC, to be superseded by the Medical Devices Regulation (MDR). It should be stated whether the Clinical Evaluation Reports is in support of initial CE-marking, a CE mark renewal, or is at the request of the Notified Body (NB). The documents required for all CERs and those additional documents specific to CE marked devices or to new devices, where equivalence with another devices is being claimed.
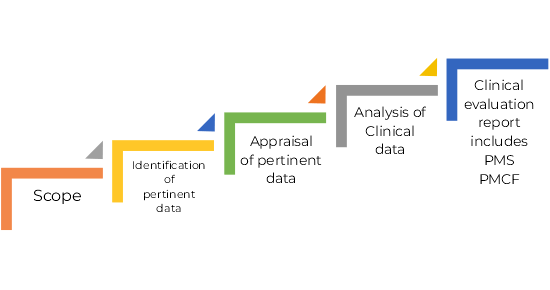
Stages of Clinical Evaluation
Stages of Clinical Evaluation
How to Obtain Accurate Data for Clinical Evaluation Reports
It is important that manufacturer should know whether they have the right data available or not;
- Manufacturer technical information.
- Clinical investigations if the device is new.
- Literature search through Databases.
- Predicate device search to prove Equivalence for device.
- Post market activities if the device is already available in market.
- Identification of risks through Risk management process in-order to claim safety of the device.
- How device achieve its intended purpose All known and foreseeable risk, any adverse event, contraindication, are minimized acceptable when weighed against the benefits of the intended use.
- That any claims made about the device’s performance and safety are supported by suitable evidence.
- Safety Precaution during use of device.
For products that are already on the market, companies should be conducting ongoing post-market surveillance to gather data on the real-world use of their devices; this identifies unforeseen risks, new contraindication, adverse event and complications, as well as areas for improvement. If they have been failing to do this, they will have to begin the time-consuming and costly process of collating data.
The roles of clinical evaluation and clinical investigation become far more prominent under the MDR, many requirements of MEDDEV and ISO 14155 are incorporated in MDR. New and tighter criteria are introduced for demonstrating equivalence. As a result, more clinical data must be obtained from clinical investigations of the device. Implantable and Class III devices generally require clinical investigations, unless a rationale can be provided for why this should not be the case. Manufacturers of implantable and Class III devices may consult an expert panel on a voluntary basis prior to the clinical evaluation. A manufacturer may rely on clinical data of another device if the new device is a modification of the old device, if the NB has confirmed this is only a modification, and if the manufacturer has full access to the technical documentation of the other device. The electronic system must also be used for PMCF studies. The design, execution, and requirements for documentation of a PMCF study have to meet many requirements applicable to clinical investigations.
Clinical evaluation report Sample
Clinical Evaluation report is a part of technical documentation and this sample contains contents for reference only. The contents are according to MEDDEV 2.7/1 revision 4 June 2016 article 61 and Annex XIV of MDR.
1. Summary (of the Report).
2. Scope of the Clinical Evaluation.
3. Clinical background, Current knowledge and State of the Art.
4. Device under Evaluation.
5. Clinical Evaluation Report Conclusion.
6. Date of Next Clinical Evaluation.
7. Qualification of the Responsible Evaluators.
8. Dates and Signatures.
9. Reference.
Clinical Evaluation Report for Medical Devices
The Clinical evaluation Report (CER) includes:
- Scope of the Clinical Evaluation.
- General Details (Manufacturer Name & Address, Responsible Person).
- Device Details (Description, Name, Models, Variants, Intended Purpose, Risk Class, CE marking status, Contraindications, Warnings, Precautions, Identification of Changes from the previous version –if already in market).
- the details of the clinical background, current knowledge, and state of the art which can be used to evaluate the safety and performance of the device for corresponding to its intended purpose.
- If a well-established ⦁ CE marked device which is similar to the device under evaluation, then the reports of that device can be used to prove the safety and performance of the device under evaluation.
- Details of the post-market activities conducted are also provided in the CER, which is used to answer any unanswered questions or residual risks that are not covered by the available clinical data.
- Tests conducted are listed to prove the safety and performance of the device.
- During the analysis of the appraised data, the requirements on safety, performance, acceptability of benefit/ risk profile, and side-effects are assessed to establish conformity to GSPR.
- Safety and performance data through literatures.
- Summary of the suitability of the device, including its IFU, for the intended users and usability aspects.
- The conclusion of.
- CER includes the acceptability of the risk-benefit profile according to current knowledge/ the state of the art in the medical fields concerned and according to available medical alternatives.
- The Clinical Evaluation Report sample is used to ultimately evaluate and prove the device is safe for use on humans and that it performs as expected when used according to the manufacturer’s instructions.
- Also, the CER shows that the presence of the device on the market is justified because of side-effects and risks, if any, are outweighed by the benefits of the device.
Maven Profcon’s Approach to Clinical Evaluation Reports
Maven Profcon Services LLP is a medical device consultancy located At Ahmedabad India focused on medical devices. We have well qualified, dedicated and experience team for conducting medical device clinical studies as per the requirement of Europe. We also help to customers for PMS & PMCF studies, Data analysis, support to customers for conformity of the device with the essential requirements for safety and performance as per the European Medical Devices Directive (MDD) 2007/47/EC, to be superseded by the Medical Devices Regulation (MDR) are require for initial CE marking or a CE mark renewal.
- Preparation of CERs as per the requirement of MEDDEV 2.7/1 revision 4.
- European Post Marketing Clinical Follow-up Studies for Medical Device.
- Search & Analysis of Clinical Data.
- Clinical Investigation as per ISO 14155 for Medical Device Clinical Studies.
- Training on, how to prepare Clinical Evaluation Reports, what are data require for preparation of CER according to MEDDEV 2.7/1 revision 4.
- Gap Analysis of CER according to MEDDEV 2.7/1 revision 4.
- Clinical Evaluation as per article 61 and Annex XIV of MDR and MDCG guidance documents.