ISO 14971 is the globally recognized standard that outlines the framework for implementing effective risk management processes throughout the lifecycle of a medical device, including Software as a Medical Device (SaMD) and In Vitro Diagnostic Devices (IVDs). It helps manufacturers identify potential hazards, assess and evaluate associated risks, implement control measures, and monitor the effectiveness of these controls. The standard plays a critical role in ensuring device safety and performance, and is often used in conjunction with ISO 13485, which mandates a quality management system for medical device manufacturers.
ISO 14971 is applicable not only to manufacturers but also to suppliers, service providers, and other stakeholders involved in the device lifecycle. The standard aligns with key regulatory requirements from global authorities like the US FDA and the European Commission under the EU MDR 2017/745. By embedding risk management into every stage of product development and post-market surveillance, ISO 14971 helps manufacturers maintain compliance, reduce liability, and enhance patient safety.
ISO 14971 specifies terminology, principles and a process for Medical Device Risk Management, including Software as Medical Device and InVitro Diagnostic Medical Devices. The process described in ISO 14971 intends to assist manufacturers of medical devices to identify the hazards associated with the medical device, to estimate and evaluate the associated risks, to control these risks, and to monitor the effectiveness of the controls.
The process described in ISO 14971 can also be applied to products that are not necessarily medical devices in some jurisdictions and can also be used by others involved in the medical device life cycle.
“Risk management can be an integral part of a quality management system”
The different stages of the Risk Management, and how they interact, remain unchanged. Nonetheless, there are still some remarkable changes, listed further below, in comparison to the previous version of ISO 14971. Given the increased attention on benefit-risk by legislators worldwide – including the EU Commission and the US FDA, it is welcoming to see that the ISO 14971:2019 further elaborates on benefit-risk when evaluating the amount of risk involved with Medical Devices. How tISO 14971 standard addresses this increased focus on benefit-risk, is described below:
The 2007-version of ISO 14971 is explicitly mentioned in ISO 13485:2016 – Medical Devices Quality Management Mystems – Requirements for regulatory purposes as the go-to document for guidance on how to apply Medical Device Risk Management principles during device realization. Taking into account the ongoing discussions on the potential convergence of the US Quality system regulation (21 CFR 820) to the widespread ISO 13485, and the upcoming Medical Device Regulation EU MDR 2017/745 requiring manufacturers to have an active Quality Management System, it is likely that this updated version will ensure that ISO 14971 remains the global standard for product Risk Management in the Medical Device industry.
Changes compared to the previous edition of ISO 14971
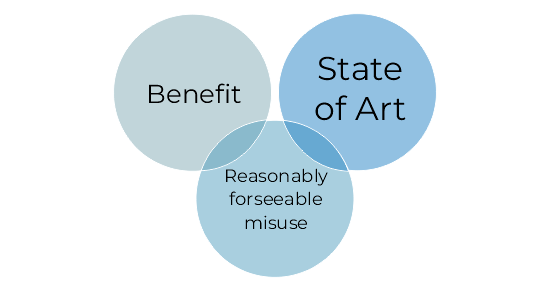
Introduction of three new definitions (benefit, reasonably foreseeable misuse & state of the art)
Increased attention to benefit-risk analysis, aligning the concept with terminology used in certain regulations, such as the MDR.
Additional emphasis on the scope of the ISO 14971-risk management process, i.e. all risks associated with a medical device, ranging from risks related to electricity, usability, data security etc.
The risk management plan has to define the methods and criteria to evaluate acceptability of the overall residual risk.
The requirements to disclose certain residual risks are merged into one requirement, as part of the “Evaluation of overall residual risk”.
More emphasis on the importance of planning of risk management activities, by stating explicitly that during risk management review the proper execution of the risk management plan has to be verified.
The requirements with regards to production and post-production activities as part of risk management have been elaborated and restructured.
The number of annexes to the standard have been decreased and the information moved to ISO/TR 24971, in order to maintain the focus on the normative requirements.
Benefit-risk: New definitions and risk-benefit reshuffle
Clause 3 in the 2019 version of ISO 14971 maps to Clause 2 in the 2007 version of ISO 14971.
Clause 5, Risk Analysis maps to Clause 4 in 2007 version of ISO 14971, is revised to give it “a more logical order.
Clause 5.1 describes the general risk management process and notes that device-makers must record risk management results in a risk management file, among other directions.
And Clause 5.2 goes into some detail about intended use and reasonably foreseeable misuse. It says “the intended use should take into account information such as the intended medical indication, patient population, part of the body or type of tissue interacted with, user profile, use environment, and operating principle.”
Clause 5.3 talks about the identification of characteristics related to safety, while Clause 5.4 focuses on the identification of hazards and hazardous situations. And Clause 5.5 is all about risk estimation.
Clause 8 – Evaluation of Overall Residual Risk, which maps to Clause 7 in the 2007 standard, “says the manufacturer must evaluate the overall residual risk. When the residual risk is unacceptable, you can do a benefit-risk analysis on the overall residual risk.
Clause 10 – Production and Post-Production Activities – This clause takes one large section in ISO 14971:2007 – Clause 9, “Production and Post-Production Information” – and restructures it into three bite-sized sections.
10.1 Information Collection 10.2 Information Review 10.3 Actions
Implementation of ISO 14971 in Practice
Implementing ISO 14971 in a real-world setting requires integration into the organization’s existing quality management system (QMS), typically ISO 13485. This begins with establishing a robust risk management framework, which includes setting up risk management policies, assigning responsibilities, and developing procedures for each stage—risk analysis, evaluation, control, and post-market surveillance. Cross-functional collaboration is essential, involving design, clinical, manufacturing, and regulatory teams. Practical tools like Failure Mode and Effects Analysis (FMEA), Fault Tree Analysis (FTA), and Hazard Analysis are commonly used to identify and mitigate risks. Documentation is a cornerstone of ISO 14971; a risk management file must be maintained throughout the product lifecycle. Additionally, post-market feedback, complaints, and vigilance reports must be routinely analyzed to update the risk assessment. Regular training, internal audits, and management reviews ensure continuous improvement and compliance. With the growing complexity of devices, especially Software as a Medical Device (SaMD), structured implementation of ISO 14971 is critical for regulatory approval and patient safety.
Common Challenges and Best Practices
Implementing ISO 14971 can pose several challenges for medical device manufacturers. One common challenge is accurately identifying all potential hazards, including those related to software, usability, and cybersecurity, which are increasingly critical in today’s connected medical devices. Another difficulty lies in performing thorough benefit-risk analyses, especially when balancing patient safety against device performance and innovation. Additionally, maintaining comprehensive documentation throughout the product lifecycle can be resource-intensive but is essential for regulatory compliance and traceability.
To overcome these challenges, best practices include early integration of risk management activities within the product development process, ensuring cross-functional collaboration between engineering, clinical, and regulatory teams. Leveraging risk management tools and software can streamline hazard identification and risk evaluation. Continuous training and updates on regulatory changes help maintain compliance with evolving standards. Finally, establishing a robust post-market surveillance system supports ongoing risk assessment and timely mitigation of emerging risks, ensuring device safety and effectiveness throughout its lifecycle.
Future Trends in Medical Device Risk Management
As the medical device industry continues to evolve rapidly, the future of risk management is expected to incorporate several emerging trends and technologies to enhance patient safety and regulatory compliance. One key trend is the increasing integration of artificial intelligence (AI) and machine learning into risk assessment processes. These technologies can analyze vast amounts of real-world data to identify potential risks earlier and more accurately than traditional methods.
Another important development is the growing emphasis on cybersecurity risk management. With medical devices becoming more connected through the Internet of Medical Things (IoMT), protecting devices from cyber threats has become a critical aspect of overall risk management.
Additionally, the shift towards personalized medicine and wearable medical devices introduces new challenges in managing risks associated with patient-specific factors and continuous data monitoring. Regulatory bodies are also focusing on harmonizing global standards, which will require manufacturers to adopt more comprehensive and adaptable risk management frameworks.
Finally, post-market surveillance is expected to play a bigger role, with real-time monitoring systems providing ongoing data to detect emerging risks, helping manufacturers to react promptly and improve device safety throughout its lifecycle.
Conclusion
The updated ISO 14971 – Medical devices – Application of risk management to medical devices does not cause a major shift in the perception of risk within the medical device industry. The current risk management process (risk analysis, risk evaluation, risk control & evaluation of residual risk) is not drastically revamped, as the authors confirmed the risk management process as defined in the previous version. However, the level of attention given to the concept of benefit, and how it compares to risk, is new in this version. Whereas the concept was already present in the previous versions of the ISO 14971, the new version is now fully embracing this concept. By doing this, the ISO 14971:2019 – Medical devices – application of risk management to medical devices clearly addresses the global requirements for risk management.
If you have any questions about the update, our experts will be happy to help you. Just send us a message and we will be in touch shortly.