The verdict is out! The ball is now in the manufacturer’s court to take adequate measures to make the best of this extension in the validity of its current CE certificate.
In continuation to the proposal put forward by the EPSCO for the extension to the transition date for Regulation (EU) 2017/745 (MDR) and Regulation (EU) 2017/746 (IVDR) at the meeting in Brussels last month, the EU commission has amended the regulation to include the suggested changes looking at the current scenario and considering public health and safety as a priority.
This blog intended to grill down these changes into a precise piece of information for manufacturers to conclude on the best possible strategies to comply and take benefit of this extension.
Amended Article 120: Relief Measures and Prerequisites for EU MDR Transition
Article 120, which has been the guiding light for all legacy devices certified under Directives 90/385/EEC and 93/42/EEC, has been modified critically to allow some relief to the manufacturers and notified bodies; however, there are prerequisites in trying to take advantage of the modifications, and the entire aim of doing so is to see serious action from the manufacturer’s end to demonstrate significant efforts in transitioning to EU MDR.
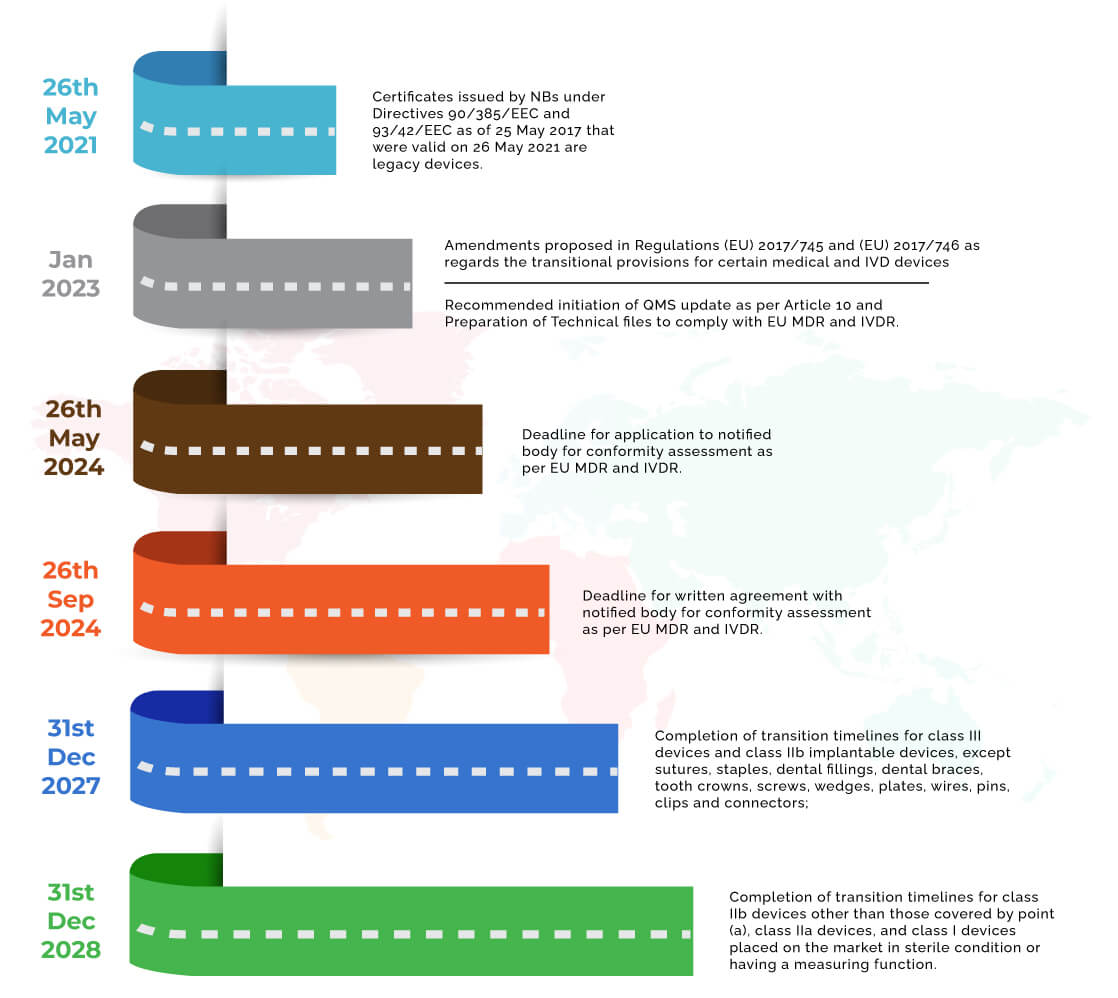
The amended Article 120 (3b) states that devices ‘Certificates issued by notified bodies under Directives 90/385/EEC and 93/42/EEC as of 25 May 2017 that were valid on 26 May 2021 and that have not been withdrawn afterwards shall remain valid after the end of the period indicated on the certificate until the following dates:
(a) 31 December 2027, for class III devices and class IIb implantable devices, except sutures, staples, dental fillings, dental braces, tooth crowns, screws, wedges, plates, wires, pins, clips and connectors;
(b) 31 December 2028, for class IIb devices other than those covered by point (a), class IIa devices, and class I devices placed on the market in sterile condition or having a measuring function.
However, it is critical to note here that the devices for which the certificate has already expired before the publication of this amended regulation can only benefit from this extension provided they have already signed an agreement with notified body before the date of expiry of the certificate.
The devices for which notified body assessment were not required under the earlier directive 93/42/EEC. For which the declaration of conformity was drawn up earlier to 26 May 2021 and has now been up and classified into a higher classification requiring notified body review as per EU MDR 2017/745 can be continued to be placed in the EU market or put into service till 31 December 2028.
Conditions for Extending Certificate Validity Under EU MDR and IVDR
These extensions come with expectations and require efforts made by the manufacturer to show significant attempts to adhere to the new regulation (EU) 2017/745 And (EU) 2017/746. The following conditions have been set to ensure the wise use of this extension and that only manufacturers who have made a genuine attempt to continue selling their devices to the EU by complying with the New Regulation (EU) 2017/745 And (EU) 2017/746.
- The devices shall comply with Directive 90/385/EEC or Directive 93/42/EEC.
- The product design & and its intended use should remain the same.
- The use of the device should not pose any significant risk or harm to the health and safety of the patient, user or any other person who may come in contact with the device or public health in general.
- An essential condition where the manufacturers have to demonstrate effort is the update their Quality Management System as per the requirements of Article 10 (9) of (EU) 2017/745 (MDR).
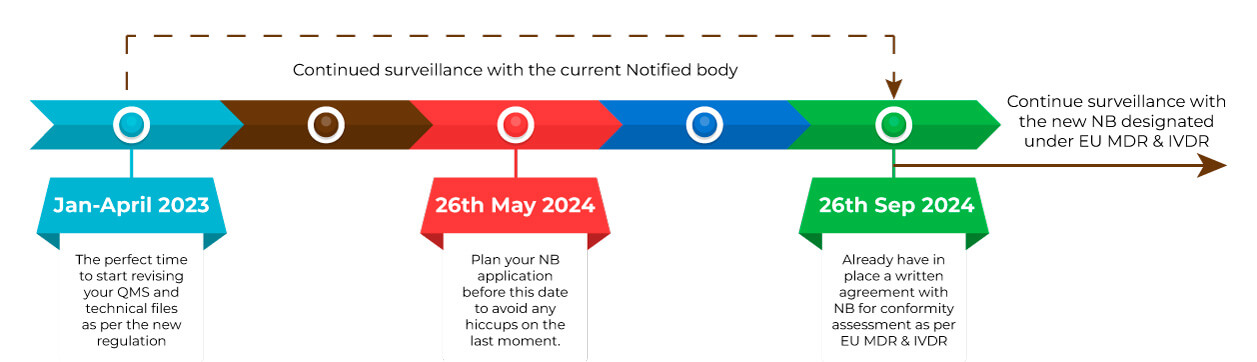
- The most significant condition is that the manufacturer of its authorised representative should have formally lodged an application to the notified body for the conformity assessment of the device under the new regulation before 26th May 2024. In continuation to the application, it is furthermore needed that both notified body and the manufacturer should have signed the written agreement before 26th September 2024.
Future Compliance Requirements Under Directives 90/385/EEC and 93/42/EEC
This extension shall be directly applicable and will not require changes to the current certificate. The devices shall be subject to surveillance by the notified body that issued the certificate and must comply with post-market surveillance, market surveillance, vigilance, and registration of economic operators and of devices. The notified body that issued the certificate shall be responsible unless the manufacturer has signed a written agreement with the new notified body designated under EU MDR by virtue of article 42 and the new notified body has agreed. However, the former notified body is only required to do so till 26th September 2024, after which the new notified body that has signed the written agreement shall be responsible for the surveillance of the devices that the written agreement has covered.
The transfer of surveillance from the notified body that issued the certificate to the notified body designated in accordance with Article 42 shall be defined in an agreement between the manufacturer, the notified body designated in accordance with Article 42 and, where practicable, the notified body that issued the certificate.
Transition for Class III custom-made implantable devices
The transition timelines for manufacturers of Class III custom-made implantable devices ended on 26th May 2021 and were already required to comply with EU MDR and were not covered by Article 120. These manufacturers, however, now need certification of their quality management system by the notified body by virtue of the conformity assessment procedure referred to in Article 52(8), and now can be continued to put in the market or service till 26th May 2026. However, it is imperative for them to have a written agreement in place with a notified body before 26th September 2024.
Key Takeaways on the Sell-Off Deadline and Next Steps for Manufacturers
The sell-off period mentioned in the current regulation has been eliminated without a legal time restriction, and devices can now be placed into use or service with no indicated limitations.
MAVEN’s Take and recommendations on the future course of action for medical device manufacturers
The EU commission looking at the current deficit in the number of notified bodies designated under the new regulations (EU) 2017/745 And (EU) 2017/746 and the heap of unviewed applications from manufacturers, has taken a significant step to ensure a sufficient number of medical devices and hence affecting patient care, diagnosis and treatment.
However, the EU commission has also ensured that these liberties be only taken by manufacturers who have made significant efforts to comply with their new regulations (EU) 2017/745 And (EU) 2017/746 and showed continued interest in providing the best quality products for the European population.
This amendment only strengthens the fact that manufacturers will now have to double up on their efforts to update their QMS, update their technical files and make applications to the notified body before 26th May 2024 to avoid any possibilities of delay or running short of notified bodies to accept their application as all manufacturers will now gear up and start doing so.
The only takeaway from this is to demonstrate continuous cooperation with your current notified body for surveillance under the former directive; meanwhile, start the process of updating your QMS and technical files to the requirements of EU MDR and consequently apply to notified bodies and come in agreement with them before the deadlines.
Critical Timelines for Compliance with EU MDR and IVDR
The crucial timelines to remember:
26th May 2024: Applications to notified bodies for conformity assessment under the EU MDR and IVDR should have been made.
26th September 2024: written agreement should have been signed with the notified body
Right now: is the time to start updating your QMS and technical files to ensure you are ready to put down application before 26th May 2024 due to the shortage in notified bodies and all certificates expiring collaterally.
While this may look like the beginning of a new race of manufacturers to start complying and chase notified bodies, we shall always remain at your disposal for any further assistance.
Author: Khushboo Chhajer