In the biological evaluation of medical devices, cytotoxicity testing is often the first formal assessment performed and for good reason. As a critical component of biocompatibility testing for medical devices, it serves as an early, sensitive indicator of whether a material or finished device may cause cellular damage when in contact with the human body.
Within the framework of ISO 10993-1, biological safety is assessed using a risk based approach. Among the various endpoints described in the ISO 10993 series, cytotoxicity defined in ISO 10993-5 is considered foundational. It provides essential data that help determine whether further biological testing is necessary and whether a device is suitable to progress toward regulatory submission as part of its overall biocompatibility evaluation strategy.
The Regulatory Significance of Cytotoxicity Testing
Cytotoxicity testing evaluates the potential of a medical device, its materials, or extractables to cause damage to cultured mammalian cells under controlled laboratory conditions. Because the test is conducted in vitro, it offers a reproducible and ethically favorable method for identifying localized toxic responses early in development.
Regulatory authorities, including the U.S. Food and Drug Administration and oversight bodies operating under the Medical Device Regulation (EU) 2017/745, expect manufacturers to address cytotoxicity as part of their biological evaluation strategy. Although the specific documentation structure may vary between jurisdictions, the scientific principles remain aligned with ISO 10993 requirements.
For most device categories regardless of contact duration cytotoxicity is considered a baseline requirement unless robust justification is provided.
Why Cytotoxicity Is Often the First Test Performed
There are several practical reasons why cytotoxicity testing is typically conducted at the early stage of biological evaluation:
- Cytotoxicity testing is sensitive to many leachable-related hazards but does not fully predict in vivo biological response.
- It has a relatively short turnaround time
- It is cost-effective compared to complex in vivo studies
- It can identify issues related to manufacturing residues, additives, or surface treatments
Because of this sensitivity, manufacturers frequently use cytotoxicity testing as a material screening tool. It provides an early indication of whether a broader suite of biological tests is likely to succeed or whether reformulation or process optimization may be required.
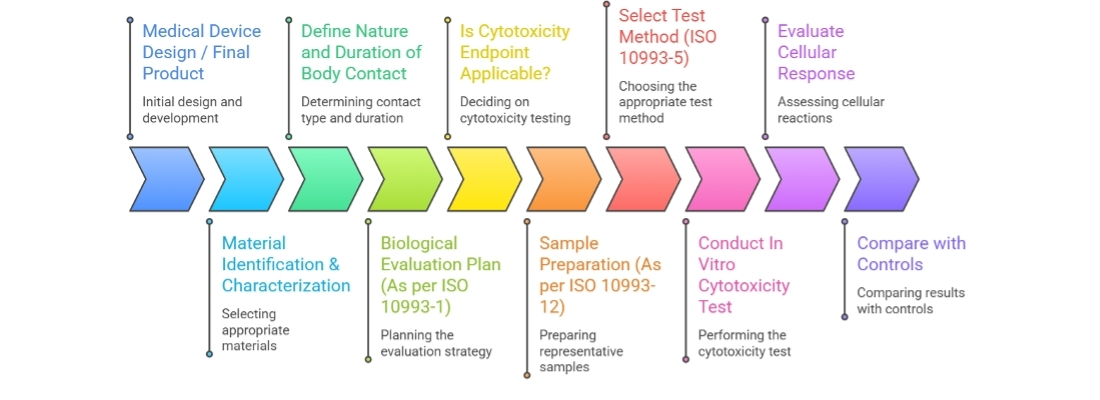
How Cytotoxicity Testing Is Performed
ISO 10993-5 describes three general approaches:
- Extract Method (Elution Test): Device extracts are prepared and applied to cultured cells.
- Direct Contact Method: The test article is placed directly onto a cell monolayer.
- Indirect Contact Method (e.g., Agar Overlay): A diffusion barrier separates the material from the cells.
Common quantitative and qualitative assays include:
- MEM Elution
- Agar Overlay
- MTT or XTT metabolic assays
- Neutral Red Uptake (NRU)
A reduction in cell viability greater than 30% (i.e., viability <70% of negative control) is considered cytotoxic.
Extraction conditions shall follow ISO 10993-12 default parameters unless scientifically justified alternatives are documented. These preparation requirements are detailed in ISO 10993-12.
Reliable cytotoxicity results depend significantly on proper sample preparation. Requirements for extraction conditions, surface area-to-volume ratios, and selection of appropriate extraction vehicles are described in ISO 10993-12.
Extraction may simulate clinical use or represent exaggerated conditions intended to reveal potential hazards. In either case, test conditions must be scientifically justified and documented.
Inadequate extraction protocols, inappropriate solvents, or unrepresentative test samples can produce misleading results. For this reason, biological evaluation must always be planned in alignment with the device’s intended use and supported by sound risk management principles.
Understanding Cytotoxicity Failures
A cytotoxicity test failure does not automatically equate to clinical risk. Instead, it signals the need for systematic investigation.
Common contributors to cytotoxic responses include:
- Residual processing chemicals
- Incomplete cleaning validation
- Sterilization by-products
- Leachable additives or colorants
- Surface modifications
When a failure occurs, manufacturers are expected to conduct root cause analysis and, where appropriate, perform chemical characterization and toxicological risk assessment. This structured evaluation ensures that decisions are based on exposure and clinical relevance rather than test outcome alone.
Integrating Cytotoxicity into a Risk-Based Strategy
The ISO 10993 framework emphasizes that biological evaluation is not a checklist exercise but a structured, risk-driven process. Cytotoxicity data must therefore be interpreted within the context of:
- Nature and duration of body contact
- Material composition
- Existing clinical history data
- Effect of Manufacturing processes
- Overall risk management documentation
When applied strategically, cytotoxicity testing supports:
- Early hazard identification
- Efficient regulatory submissions
- Reduced likelihood of redesign
- Stronger justification in Biological Evaluation Reports
Conclusion
Cytotoxicity testing represents more than a regulatory formality. It is a critical safeguard in medical device development and a central element of demonstrating compliance with ISO 10993 requirements.
By aligning cytotoxicity evaluation with ISO 10993-1, ISO 10993-5, and ISO 10993-12, manufacturers can ensure that biological safety assessments are scientifically robust, regulator-ready, and proportionate to actual patient risk.
In a regulatory environment increasingly focused on risk-based justification and data integrity, cytotoxicity remains the first and most essential checkpoint in proving medical device biocompatibility.
Reference:
ISO 10993-5:2009 – Biological evaluation of medical devices — Part 5: Tests for in vitro cytotoxicity
TÜV SÜD – Cytotoxicity Testing Overview