Introduction to Non-Conformities and Deviations
In the dynamic and highly regulated environment of medical device manufacturing, encountering non-conformities and deviations is not uncommon. Despite having a well-structured and ISO 13485:2016-compliant Medical Device Quality Management System (MD-QMS), unexpected issues can still arise due to human error, equipment malfunction, process variation, or external factors. These deviations may affect product quality, patient safety, or regulatory compliance if not handled effectively.
Understanding the nature of non-conformities—whether they are minor or major, isolated or systemic—is essential for implementing the right response. It is not just about fixing a problem when it occurs, but about identifying its cause and preventing future occurrences. Equally important is the recognition and handling of deviations, which may be planned or unplanned departures from standard procedures or specifications. A proactive approach to managing non-conformities and deviations enhances operational control, strengthens the QMS, and ultimately supports continuous improvement and patient safety—key goals in the medical device industry.
Understanding ISO 13485:2016 Guidance
ISO 13485:2016 is a globally recognized standard that provides a robust framework for establishing and maintaining a quality management system (QMS) specific to the medical device industry. One of the key strengths of the standard is its emphasis on risk-based thinking, continuous improvement, and regulatory alignment. It helps organizations proactively identify potential issues and implement effective solutions before they escalate into critical problems.
The standard doesn’t just stop at compliance—it pushes organizations to adopt a culture of accountability and traceability. This means every action taken—be it a correction, corrective action, or preventive action—must be backed by documented evidence and evaluated for effectiveness.
Moreover, ISO 13485 encourages a structured approach to handling feedback, complaints, and audits, making it easier for companies to maintain product safety, meet customer expectations, and uphold regulatory commitments. Understanding and applying this guidance properly helps create a resilient and high-performing MD-QMS.
Difference Between Correction, Corrective Action, and Preventive Action
Understanding the difference between correction, corrective action, and preventive action is not just about regulatory compliance—it’s about cultivating a culture of quality and continuous improvement. Many organizations mistakenly treat these terms interchangeably, leading to ineffective responses and missed opportunities to improve the system. Corrections are reactive—solving the problem temporarily without altering the system. Corrective actions go deeper, addressing root causes to ensure similar issues do not recur. Preventive actions, on the other hand, are proactive—they rely on risk assessment, trend analysis, and foresight to eliminate potential issues before they arise.
For medical device manufacturers, proper documentation and implementation of these actions are essential during audits and inspections. Regulatory bodies like the FDA or notified bodies under the EU MDR closely evaluate how companies manage non-conformities and potential risks. Failure to distinguish these actions correctly could signal a weak QMS and may result in non-compliance findings. A robust, well-differentiated CAPA (Corrective and Preventive Action) system is a cornerstone of quality assurance and patient safety.
As medical device manufacturers, it is quite usual that you come across non-conformities and other deviations in the product or process within your Medical Device Quality Management System (MD-QMS). Not that we want it to happen, but a well-established MD-QMS doesn’t necessarily guarantee ZERO deviations. After all, to err is human!
However, we sure should know how to deal with deviations to correct, avoid and prevent its occurrence and recurrence. There seems to be a bit of confusion that persists on the understanding of the different types of actions that has to be taken and documented in response to a particular problem within the MD-QMS.
Thankfully, the ISO 13485:2016 standard provides us with the appropriate guidance on how to deal with such issues and come up with an effective solution to address these deviations. Let us have a look at what the standard has to say:
CORRECTION
“The immediate action to correct a problem”.
Although not quite clearly defined in the standard as such, a CORRECTION is a short-time, one-step solution to a particular non-conformity/observed deviation/problem. Please take a look at the example below;
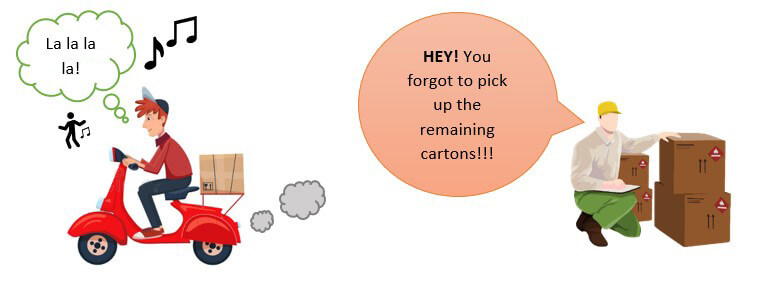
Suppose that you are medical device manufacturer who has to supply 100 units of the medical device to your customer. But due to human error, the delivery assistant missed out to pick up the whole lot and delivered only 50 units to the customer!
Very well, your customer would get upset with this mistake on your behalf and raise a complaint regarding the missing number of products.
What would be the CORRECTION here?
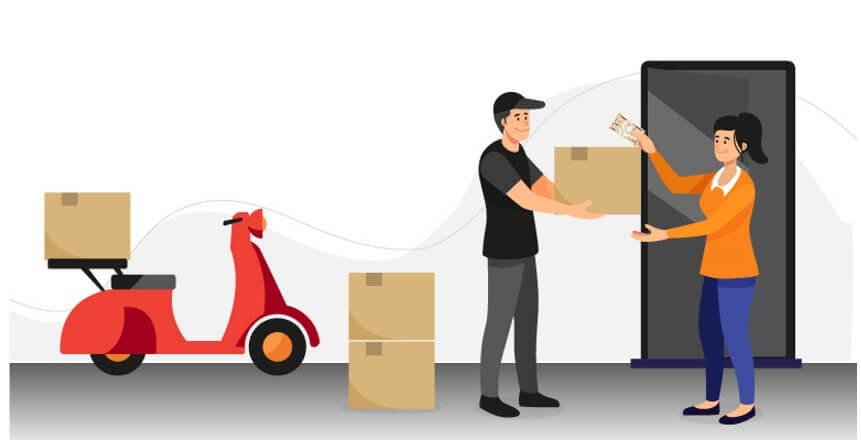
Call up the customer, apologize for the inconvenience caused and make arrangements for the dispatch of the remaining products at the earliest. Easy!
It is important to note that a correction will NOT impact the MD-QMS in any way, whatsoever.
CORRECTIVE ACTION
“The organization shall take action to eliminate the cause of nonconformities in order to prevent RECURRENCE.”
As stated in Clause 8.5.2 of the standard “evaluating the need for action to ensure that nonconformities do not recur” is a corrective action. This can only happen if and only if you identify what caused the problem in the first place… i.e., perform a root cause analysis and identify the ROOT CAUSE.
In continuation with the previous example, you would need to brainstorm and identify why the whole batch of products were not delivered to the customer in time.
ROOT CAUSE: After performing the root cause analysis, you came to know that there is no rechecking/double-checking activity performed by the Dispatch Supervisor before the actual dispatch.
Good going, so far! Once you have identified the root cause, you have to take the appropriate action to eliminate it. If the root cause is eradicated, the problem won’t happen again.
CORRECTIVE ACTION: Update the roles and responsibilities of the Dispatch Supervisor, update the documented procedure for dispatch, and conduct a training of your Dispatch Supervisor on double-checking the packaged units, in line with the customer order specifications before dispatch.
Please note that a corrective action will impact the MD-QMS in a major way or in a minor way (due to updates in the documentation), depending upon the nature of the occurred non-conformity against which it is taken.
PREVENTIVE ACTION
“The organization shall determine action to eliminate the causes of potential nonconformities in order to prevent their OCCURRENCE.”
Clause 8.5.3 of the standard states that any action taken to prevent the mistake from happening for the first time is a preventive action. Sounds a little strange, doesn’t it!? How can you possibly know about a mistake if it has not yet occurred!?
I shall explain it using the previous example.
The manufacturer sure did solve the problem in the Dispatch Department by taking the appropriate corrective action. But what if the same type of problem could occur elsewhere? Maybe in some other department?!?
POTENTIAL OCCURRENCE: The manufacturer, after brainstorming, rightly found out that the Quality Control Department may encounter this problem while sending samples for external lab testing, especially when there is a pre-determined number of samples to be sent.
PREVENTIVE ACTION: So, even before the problem would actually occur in the QC department and even before a query is raised by the external testing lab regarding the insufficient number of samples received, the manufacturer updated the roles and responsibilities of the QC Inspector, updated the documented procedure for quality control, and conducted a training of the QC Inspector on double-checking the number of samples sent for testing, in line with the pre-determined requirement of the lab.
And voila! You successfully prevented the problem from occurring for the first time!
Please note that a preventive action will impact the MD-QMS in a major way or in a minor way (due to updates in the documentation), depending upon the nature of the potential non-conformity against which it is taken.
Implementation Process
The successful implementation of correction, corrective, and preventive actions (CAPA) in a Medical Device Quality Management System (MD-QMS) requires a structured approach. It begins with clear identification and documentation of the issue, followed by a thorough risk assessment to understand its potential impact. Organizations must then assign responsibilities to relevant personnel, establish timelines, and ensure that all actions are traceable and verifiable. Root cause analysis tools like the 5 Whys, Fishbone Diagram, or FMEA (Failure Mode and Effects Analysis) can be effectively used to pinpoint the underlying causes. Additionally, it is crucial to communicate the planned actions across relevant departments and integrate the changes into the existing processes and documentation. Post-implementation, verification of effectiveness (VOE) must be conducted to ensure the actions taken have truly addressed the issue. Regular audits and management reviews further reinforce the effectiveness of the CAPA system and promote a culture of continuous improvement throughout the organization.
Impact on the MD-QMS
Every action—whether a correction, corrective action, or preventive action—has an impact on the Medical Device Quality Management System (MD-QMS), either directly or indirectly. While corrections are typically immediate and do not alter the structure or documentation of the MD-QMS, corrective and preventive actions often require updates to procedures, records, roles, and responsibilities. These changes help in improving the robustness of the system by addressing systemic issues or potential risks. Over time, such actions contribute to continuous improvement, enhance compliance with ISO 13485:2016, and reduce the likelihood of regulatory penalties or product recalls. Furthermore, regular monitoring of these actions through internal audits, management reviews, and CAPA logs helps maintain control and ensures that the MD-QMS evolves with the organization’s operational needs. A proactive approach in identifying and addressing root causes and potential risks not only improves product quality and customer satisfaction but also strengthens the organization’s credibility and market reputation.
CONCLUSION
A firm understanding of the different types of actions that need to be taken against received non-conformity/complaint/deviation/feedback is essential for any organization with an implemented MD-QMS. For medical device manufacturers, it is all the more crucial to remain alert and ensure that all the corrections/corrective actions/preventive actions taken address the targeted issue and close them in the best possible manner, thereby contributing to better health and happiness!